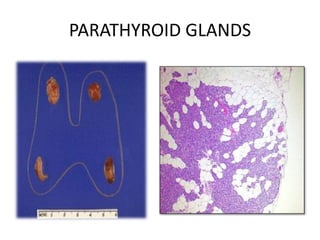
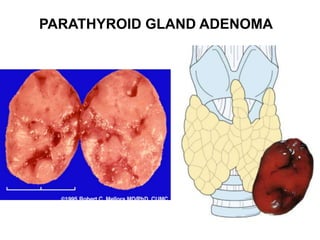
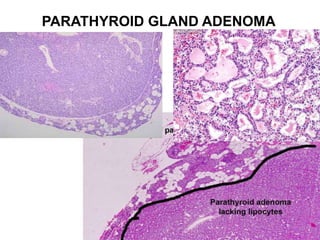
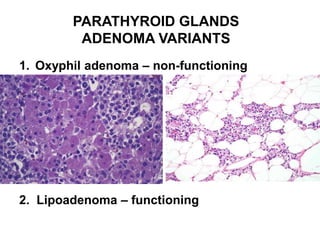
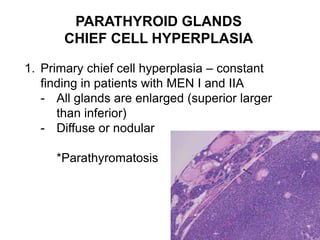
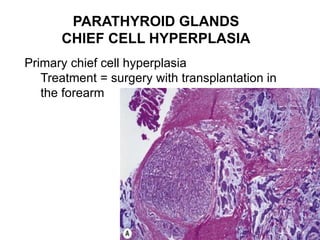
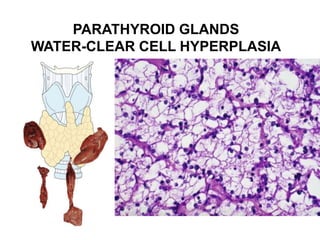
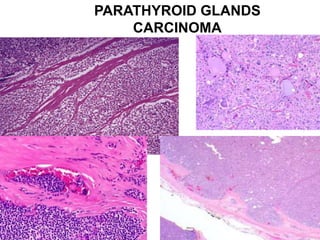
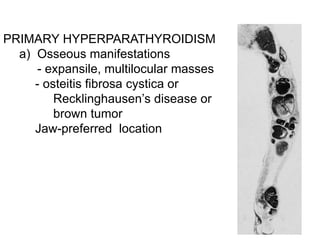
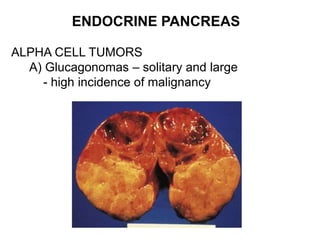
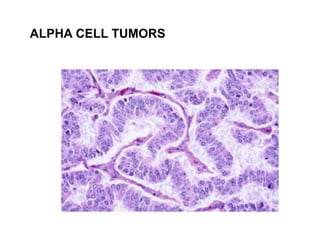
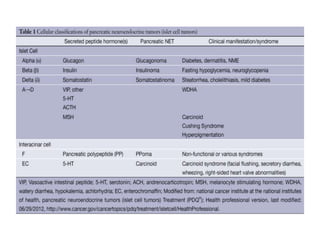
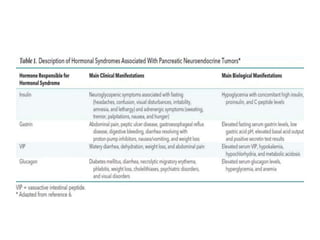
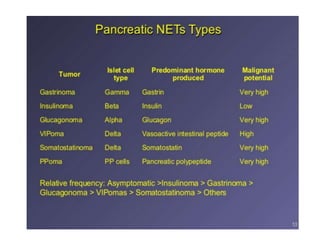
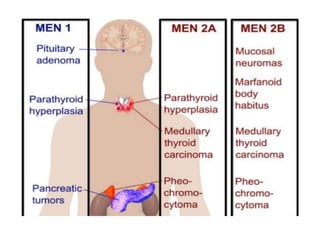
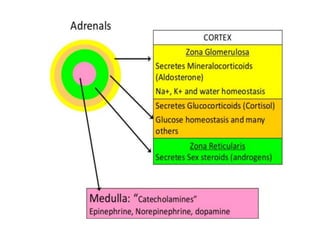
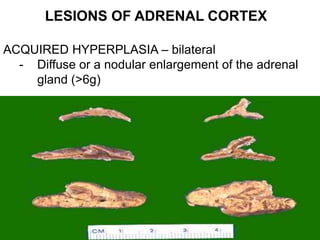
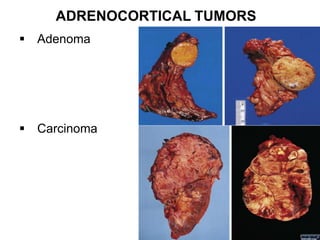
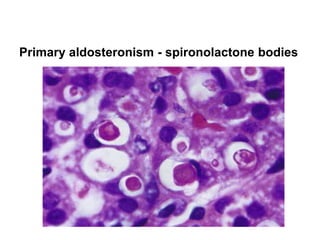
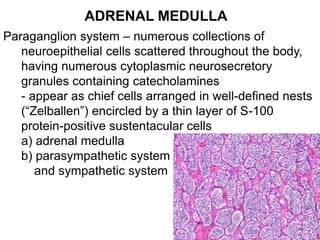
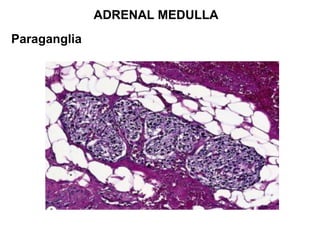
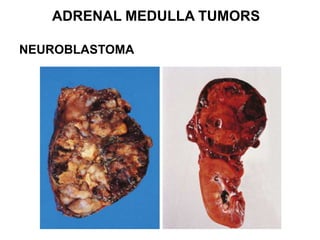
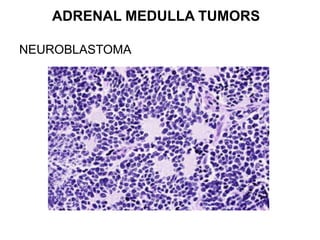
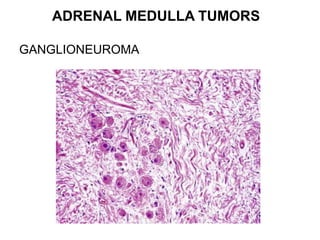
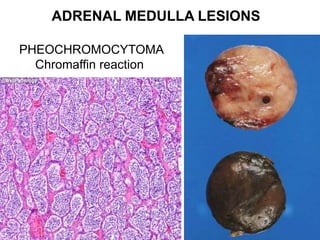
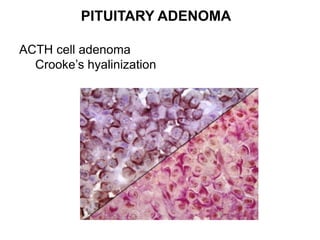
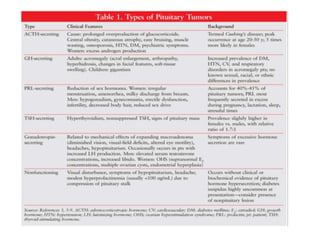
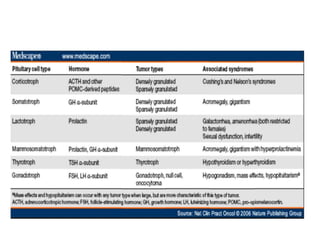
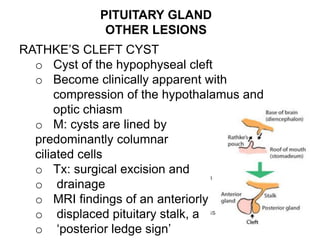
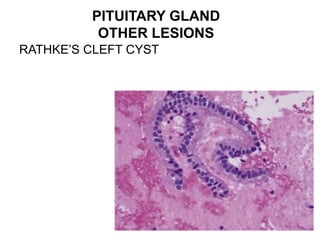
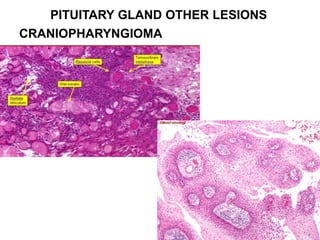
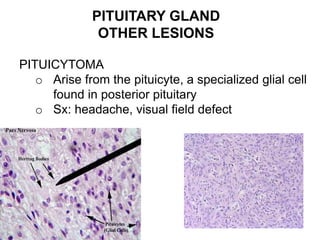
This document provides an overview of the endocrine system, focusing on the histology and pathology of key endocrine organs, including the parathyroid glands, pancreas, adrenal glands, and pituitary gland. Key lesions discussed include parathyroid adenomas, chief cell hyperplasia, pancreatic islet cell tumors, adrenal cortical lesions, and multiple endocrine neoplasia. The roles of these organs in hormone production and regulation are also reviewed.