The document outlines the features and functionalities of the accessible high throughput computing system called JIP. It details job management commands, the execution of scripts using various programming languages, and provides guidelines for creating pipelines and handling jobs. Additional resources, usage examples, and documentation links are included to assist users in implementing the system effectively.






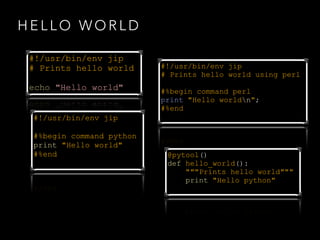
![#%begin command [perl|RScript|…]
• command block to run scripts
• specify an interpreter (default bash)
• use templates to access options and variables
#%end](https://image.slidesharecdn.com/jipintro-140116093814-phpapp01/85/JIP-Pipeline-System-Introduction-8-320.jpg)





![BED 2 BIGWIG
#!/usr/bin/env jip
# Delegates to gem-2-bed to create BED graphs from .map files
#
# Usage:
#
bed2wig -g <graph> -s <sizes> [-o <output>]
#
# Inputs:
#
-g, --graph <graph> The graph file generated with gem-2-bed
#
-s, --sizes <sizes> The sizes file generated with gem-2-wig
#
# Outputs:
#
-o, --output <output> The output file name
#
[default: ${graph|ext}.bw]
!
#%begin init
add_output('output', '${graph|name|ext}.bw')
#%end
!
#%begin setup
profile.threads = 1
#%end
!
${__file__|parent}/bedGraphToBigWig ${graph} ${sizes} ${output}](https://image.slidesharecdn.com/jipintro-140116093814-phpapp01/85/JIP-Pipeline-System-Introduction-15-320.jpg)


















































































