Downloaded 382 times




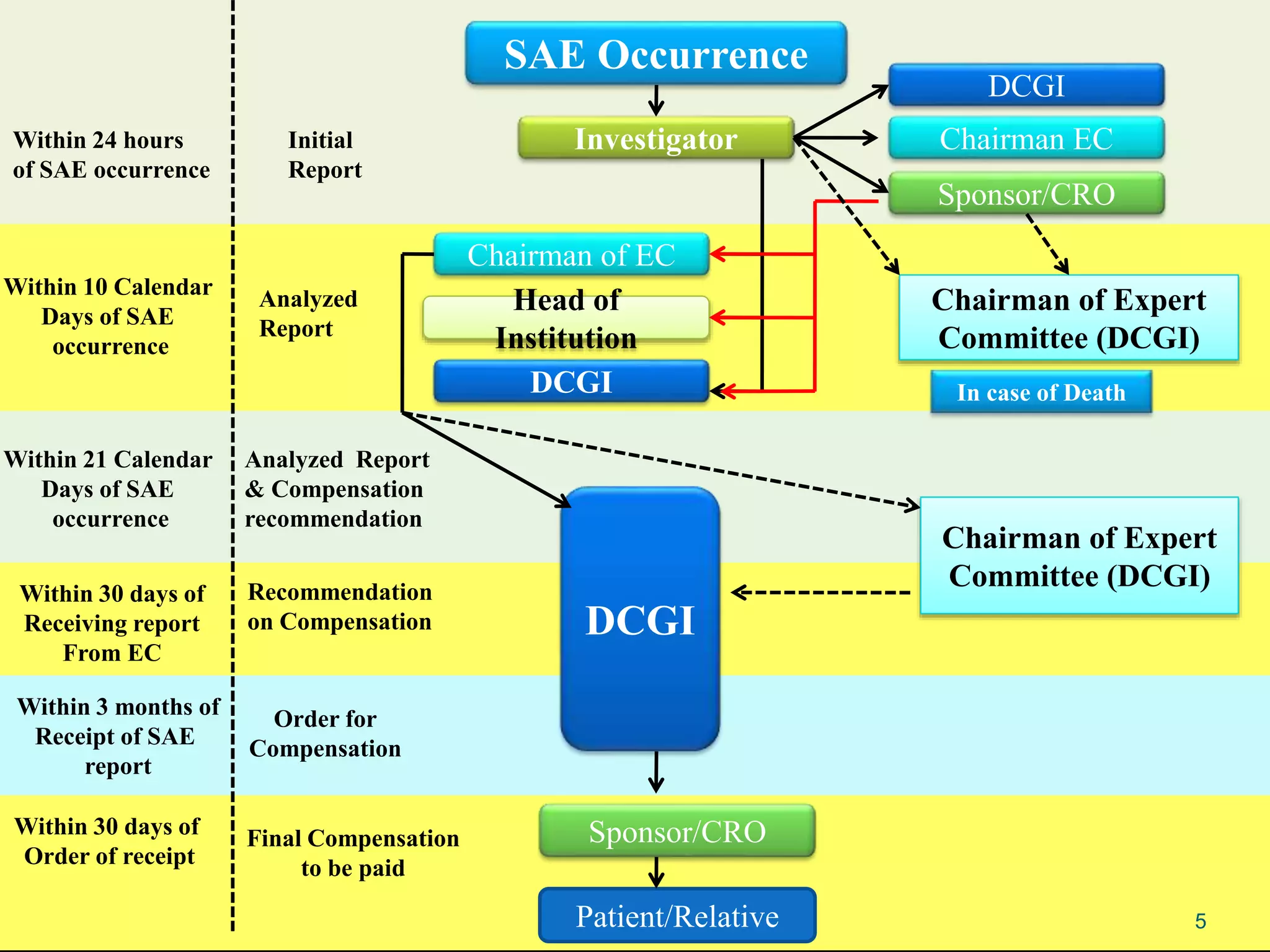




















This document provides guidance on reporting serious adverse events (SAEs) that occur during clinical trials in India. It outlines the key stakeholders involved in SAE reporting, including the investigator, sponsor/CRO, ethics committee, and Drug Controller General of India (DCGI). The timelines and process for initial and follow-up SAE reporting are defined. Requirements for the documents to be submitted in SAE reports, such as the checklist, case report form pages, and discharge summary, are also reviewed to ensure completeness and accuracy of the reports. Common errors made in SAE documentation are highlighted.















































