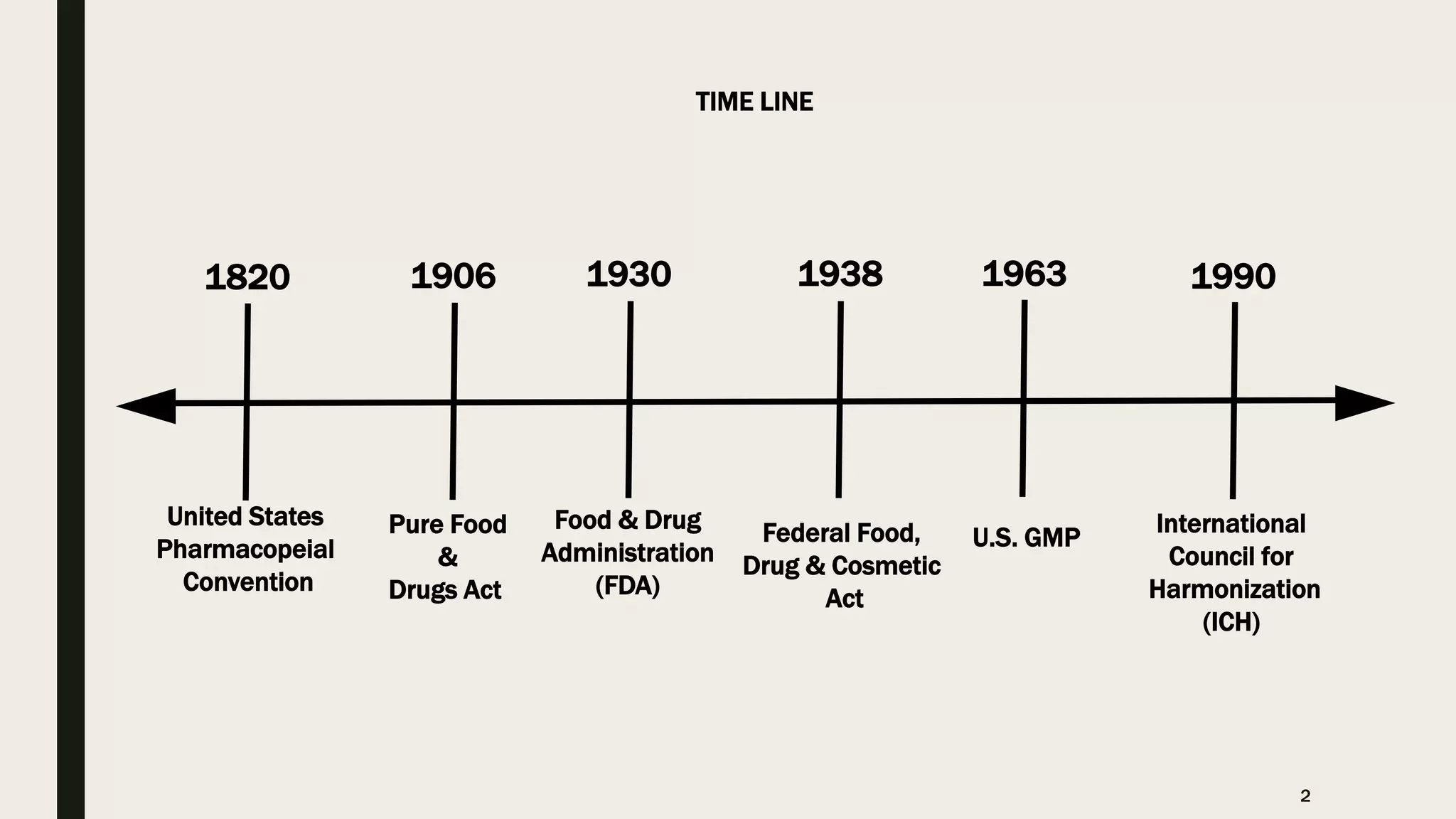
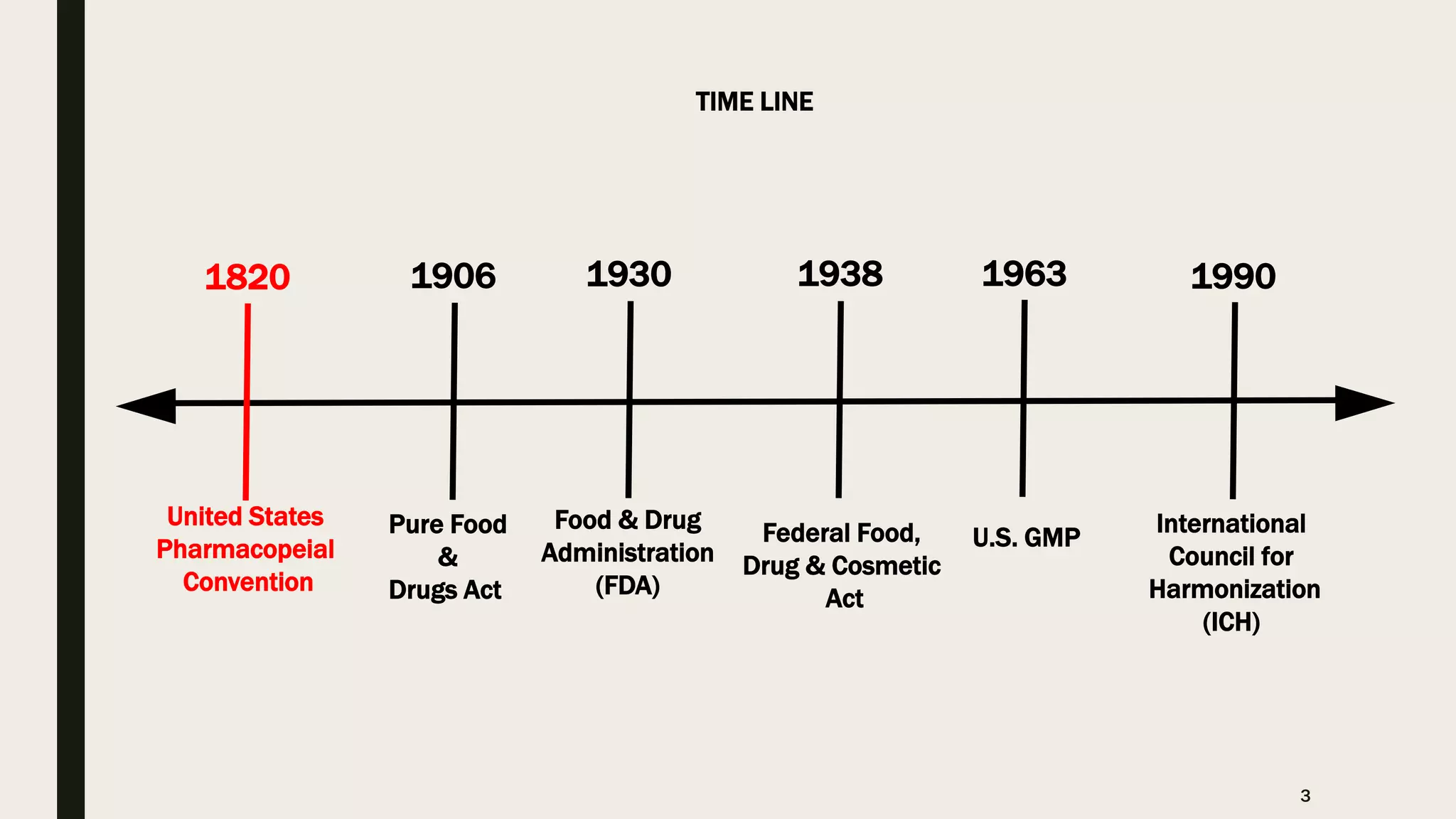
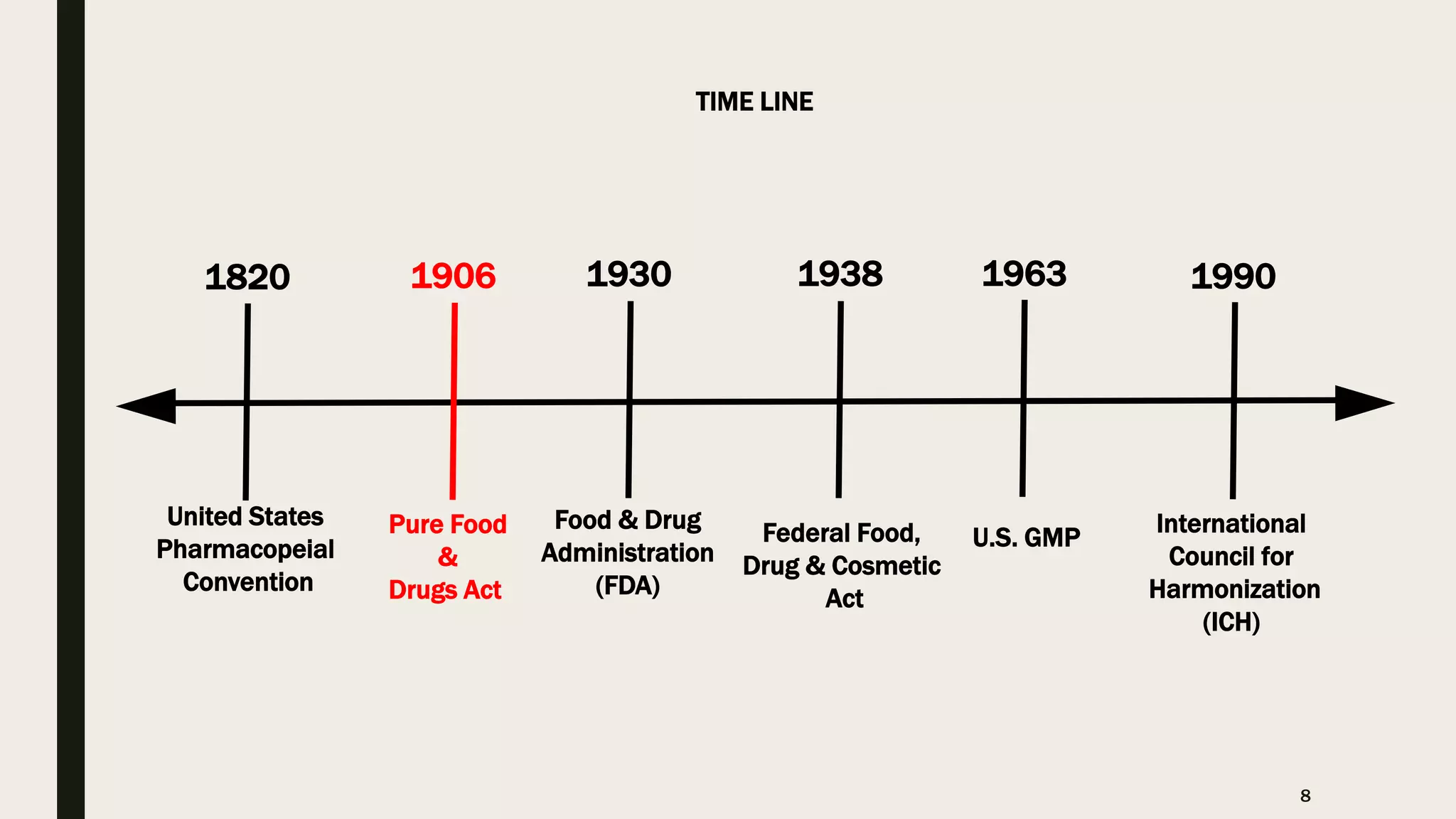
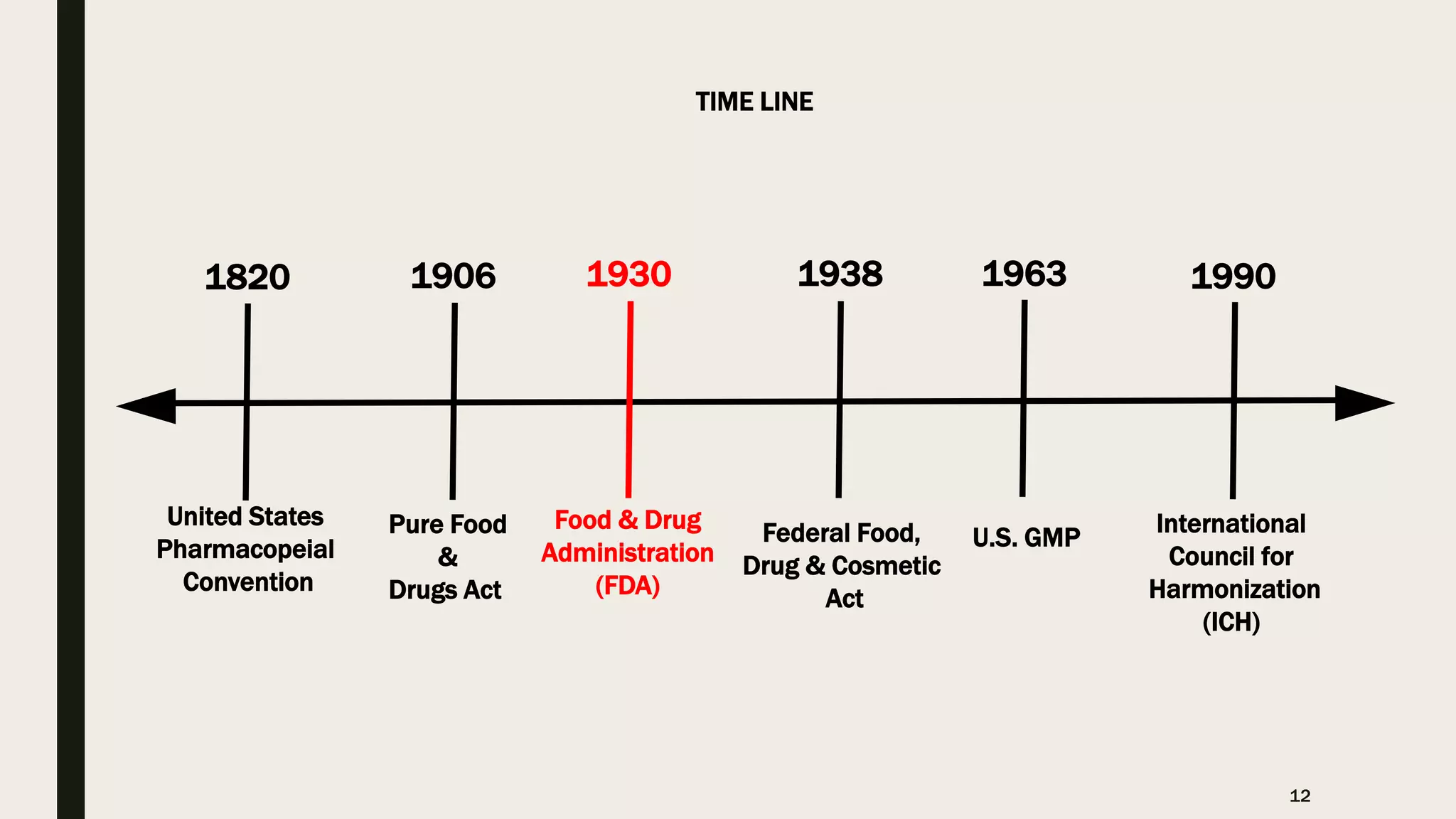
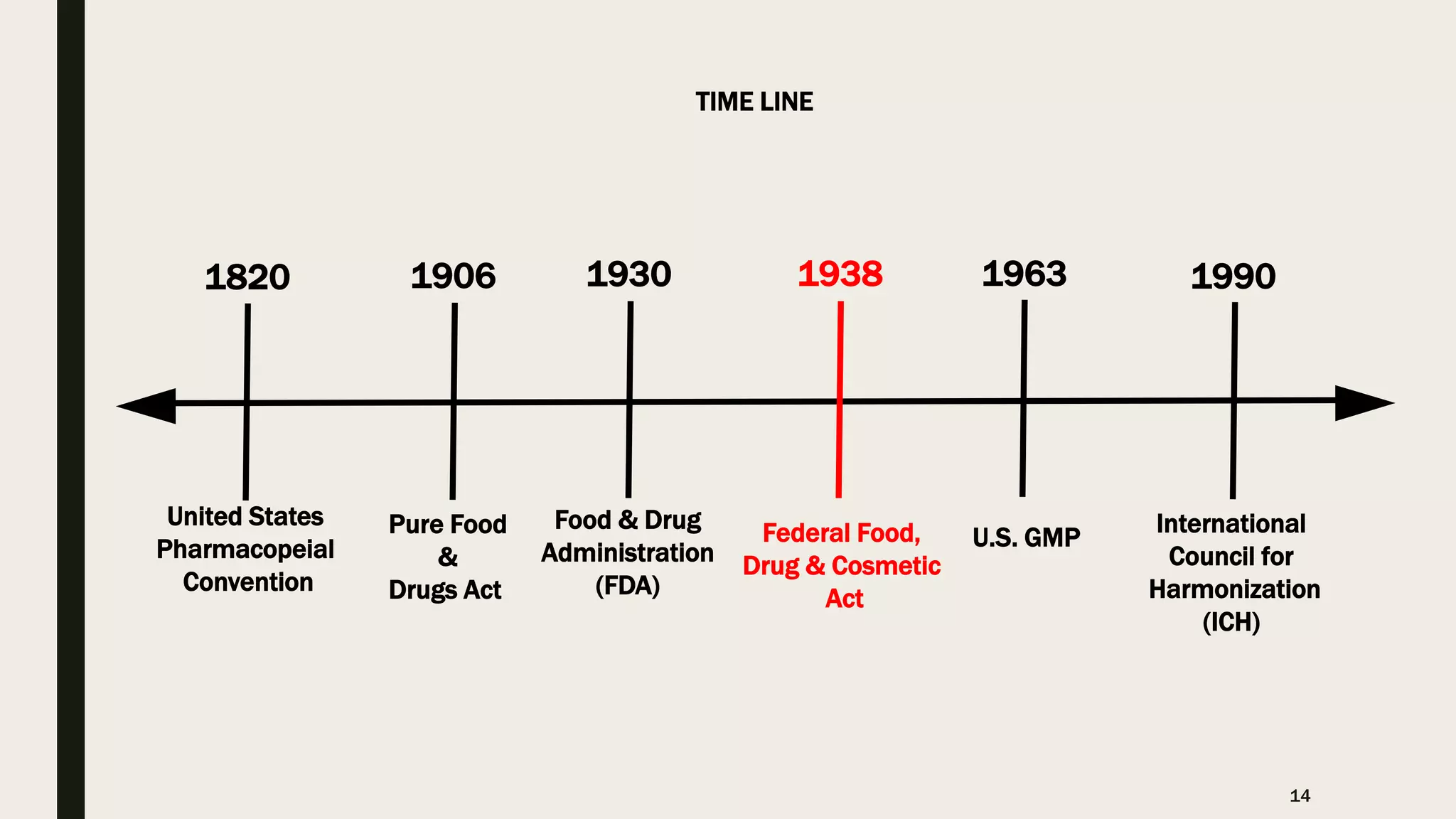
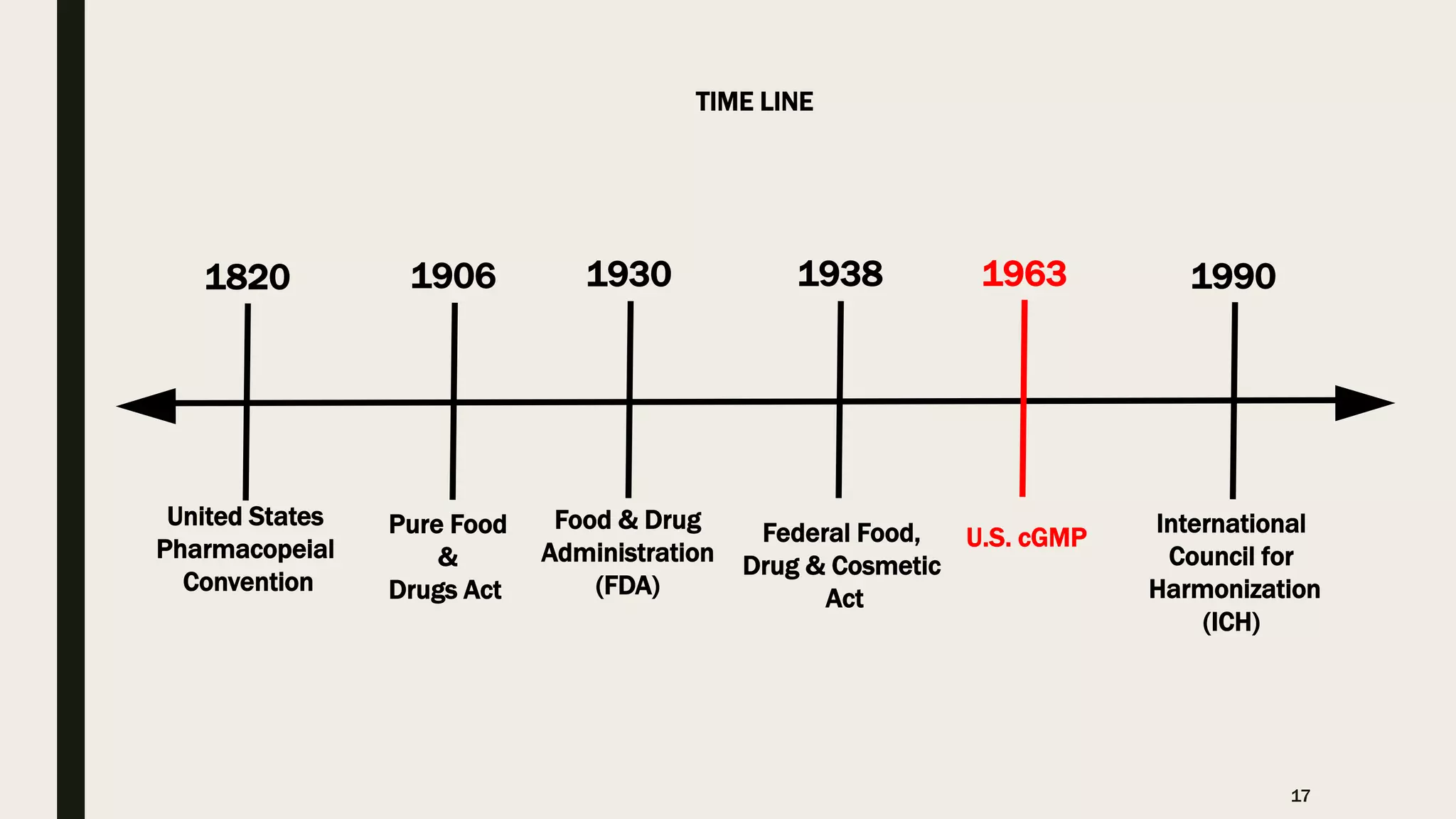
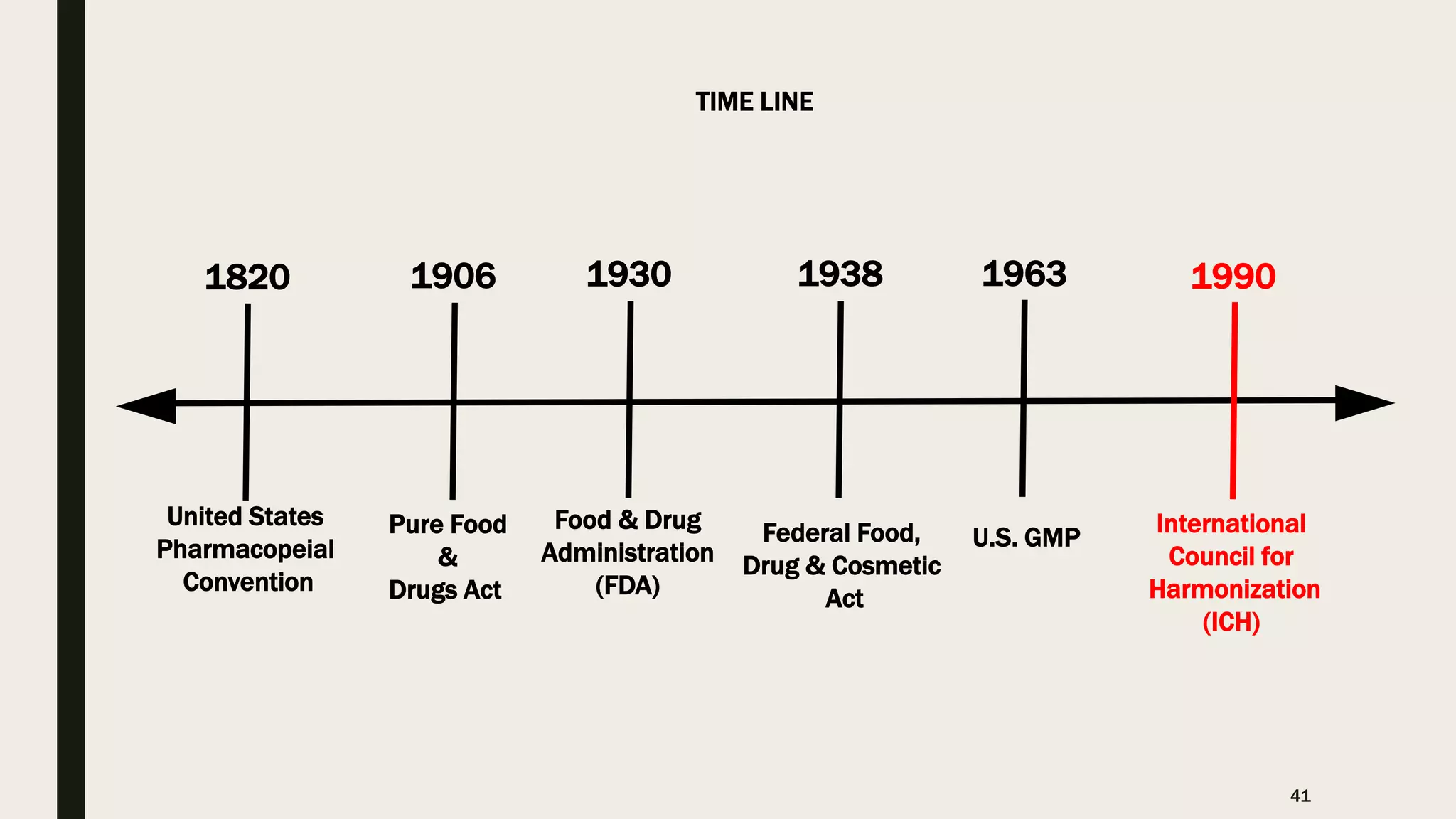
The document provides an overview of the history and principles of Current Good Manufacturing Practices (CGMP) as outlined in 21 CFR parts 210 and 211, which govern the manufacturing of pharmaceuticals in the United States. It discusses key legislation including the Pure Food and Drug Act of 1906, the Federal Food, Drug, and Cosmetic Act of 1938, and the establishment of the FDA, highlighting the evolution of regulations aimed at ensuring drug safety and efficacy. Furthermore, it emphasizes the collaborative efforts among various organizations, including the United States Pharmacopeial Convention and the International Council for Harmonisation, to harmonize pharmaceutical regulations globally.