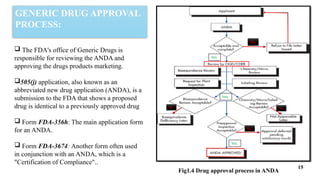
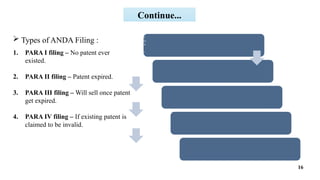
A detailed ppt on Generic drug development useful for D.pharm, B.pharm and M.pharm Students
This ppt provides brief summary to "introduction to generic drug development process", "hatch-waxman act", "steps involved in generic drug development", "ANDA filing for seeking approval of generic drug".