Download free for 30 days
Sign in
Upload
Language (EN)
Support
Business
Mobile
Social Media
Marketing
Technology
Art & Photos
Career
Design
Education
Presentations & Public Speaking
Government & Nonprofit
Healthcare
Internet
Law
Leadership & Management
Automotive
Engineering
Software
Recruiting & HR
Retail
Sales
Services
Science
Small Business & Entrepreneurship
Food
Environment
Economy & Finance
Data & Analytics
Investor Relations
Sports
Spiritual
News & Politics
Travel
Self Improvement
Real Estate
Entertainment & Humor
Health & Medicine
Devices & Hardware
Lifestyle
Change Language
Language
English
Español
Português
Français
Deutsche
Cancel
Save
Submit search
EN
Uploaded by
Amelieff
20,935 views
フリーソフトではじめるChIP-seq解析_第40回勉強会資料
2014年12月18日に開催した、アメリエフ株式会社・第40回勉強会「フリーソフトではじめるChIP-seq&メチル化データ解析入門」のChIP-Seq編のスライドです。
Health & Medicine
◦
Read more
5
Save
Share
Embed
Embed presentation
1
/ 36
2
/ 36
3
/ 36
4
/ 36
Most read
5
/ 36
6
/ 36
7
/ 36
8
/ 36
9
/ 36
10
/ 36
11
/ 36
Most read
12
/ 36
13
/ 36
Most read
14
/ 36
15
/ 36
16
/ 36
17
/ 36
18
/ 36
19
/ 36
20
/ 36
21
/ 36
22
/ 36
23
/ 36
24
/ 36
25
/ 36
26
/ 36
27
/ 36
28
/ 36
29
/ 36
30
/ 36
31
/ 36
32
/ 36
33
/ 36
34
/ 36
35
/ 36
36
/ 36
More Related Content
PDF
フリーソフトではじめるメチル化データ解析入門 SeqCap Epiデータ対応_第40回勉強会資料
by
Amelieff
PPTX
【DL輪読会】時系列予測 Transfomers の精度向上手法
by
Deep Learning JP
PDF
生存時間分析の書き方
by
Yasuyuki Okumura
PDF
機械学習と主成分分析
by
Katsuhiro Morishita
PPTX
分割時系列解析(ITS)の入門
by
Koichiro Gibo
PDF
PFI Seminar 2012/03/15 カーネルとハッシュの機械学習
by
Preferred Networks
PDF
フリーソフトで始めるNGS解析_第41・42回勉強会資料
by
Amelieff
PDF
機械学習のためのベイズ最適化入門
by
hoxo_m
フリーソフトではじめるメチル化データ解析入門 SeqCap Epiデータ対応_第40回勉強会資料
by
Amelieff
【DL輪読会】時系列予測 Transfomers の精度向上手法
by
Deep Learning JP
生存時間分析の書き方
by
Yasuyuki Okumura
機械学習と主成分分析
by
Katsuhiro Morishita
分割時系列解析(ITS)の入門
by
Koichiro Gibo
PFI Seminar 2012/03/15 カーネルとハッシュの機械学習
by
Preferred Networks
フリーソフトで始めるNGS解析_第41・42回勉強会資料
by
Amelieff
機械学習のためのベイズ最適化入門
by
hoxo_m
What's hot
PDF
正準相関分析
by
Akisato Kimura
PPTX
(実験心理学徒だけど)一般化線形混合モデルを使ってみた
by
Takashi Yamane
PDF
学位論文の書き方メモ (Tips for writing thesis)
by
Nobuyuki Umetani
PPTX
Self-Critical Sequence Training for Image Captioning (関東CV勉強会 CVPR 2017 読み会)
by
Yoshitaka Ushiku
PPTX
Go-ICP: グローバル最適(Globally optimal) なICPの解説
by
Yusuke Sekikawa
PDF
POMDP下での強化学習の基礎と応用
by
Yasunori Ozaki
PDF
SNPデータ解析入門
by
Amelieff
PDF
論文紹介-Multi-Objective Deep Reinforcement Learning
by
Shunta Nomura
PDF
Stan超初心者入門
by
Hiroshi Shimizu
PPTX
【DL輪読会】Towards Understanding Ensemble, Knowledge Distillation and Self-Distil...
by
Deep Learning JP
PDF
一般化線形モデル (GLM) & 一般化加法モデル(GAM)
by
Deep Learning Lab(ディープラーニング・ラボ)
PDF
変分推論と Normalizing Flow
by
Akihiro Nitta
PPTX
fastTextの実装を見てみた
by
Yoshihiko Shiraki
PDF
Lucas kanade法について
by
Hitoshi Nishimura
PDF
工学系大学4年生のための論文の読み方
by
ychtanaka
PDF
ブースティング入門
by
Retrieva inc.
PDF
ドメイン適応の原理と応用
by
Yoshitaka Ushiku
PDF
混合モデルを使って反復測定分散分析をする
by
Masaru Tokuoka
PPTX
Dimensionality reduction with t-SNE(Rtsne) and UMAP(uwot) using R packages.
by
Satoshi Kato
PPTX
ようやく分かった!最尤推定とベイズ推定
by
Akira Masuda
正準相関分析
by
Akisato Kimura
(実験心理学徒だけど)一般化線形混合モデルを使ってみた
by
Takashi Yamane
学位論文の書き方メモ (Tips for writing thesis)
by
Nobuyuki Umetani
Self-Critical Sequence Training for Image Captioning (関東CV勉強会 CVPR 2017 読み会)
by
Yoshitaka Ushiku
Go-ICP: グローバル最適(Globally optimal) なICPの解説
by
Yusuke Sekikawa
POMDP下での強化学習の基礎と応用
by
Yasunori Ozaki
SNPデータ解析入門
by
Amelieff
論文紹介-Multi-Objective Deep Reinforcement Learning
by
Shunta Nomura
Stan超初心者入門
by
Hiroshi Shimizu
【DL輪読会】Towards Understanding Ensemble, Knowledge Distillation and Self-Distil...
by
Deep Learning JP
一般化線形モデル (GLM) & 一般化加法モデル(GAM)
by
Deep Learning Lab(ディープラーニング・ラボ)
変分推論と Normalizing Flow
by
Akihiro Nitta
fastTextの実装を見てみた
by
Yoshihiko Shiraki
Lucas kanade法について
by
Hitoshi Nishimura
工学系大学4年生のための論文の読み方
by
ychtanaka
ブースティング入門
by
Retrieva inc.
ドメイン適応の原理と応用
by
Yoshitaka Ushiku
混合モデルを使って反復測定分散分析をする
by
Masaru Tokuoka
Dimensionality reduction with t-SNE(Rtsne) and UMAP(uwot) using R packages.
by
Satoshi Kato
ようやく分かった!最尤推定とベイズ推定
by
Akira Masuda
Similar to フリーソフトではじめるChIP-seq解析_第40回勉強会資料
PDF
フリーソフトではじめるがん体細胞変異解析入門 第33回勉強会資料
by
Amelieff
PDF
NGS現場の会第2回_アメリエフ株式会社_がんExome解析
by
Amelieff
PDF
Exome解析入門
by
Amelieff
PDF
フリーソフトではじめるNGS融合遺伝子解析入門
by
Amelieff
PPTX
2014年度 第5回バイオインフォマティクス実習
by
Jun Nakabayashi
PDF
CBI学会2013チュートリアル NGSデータ解析入門 (解析編)配布資料
by
Genaris Omics, Inc.
PDF
RNASkim
by
弘毅 露崎
PDF
XCMSを用いた質量分析データ処理_BioCAsia2021_yamamoto.pdf
by
h_yama2396
PPTX
2019年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
PPTX
BGI Webinar Aug 28, 2014 "Genome wide methylation analysis and analytics"
by
kazuoishii20
PDF
NGS現場の会第2回_アメリエフ株式会社_RNAseq解析
by
Amelieff
PPTX
2016年度 第4回バイオインフォマティクス実習
by
Jun Nakabayashi
PDF
20160324自由集会講演
by
astanabe
PDF
新規医療開発に関わる統計学 (バイオインフォマティクス)
by
Hidemasa Bono
PPTX
2018年度 第1回バイオインフォマティクス実習
by
Jun Nakabayashi
PPTX
2017年度 第1回バイオインフォマティクス実習
by
Jun Nakabayashi
PPT
Kashiwa.R#9 Rでゲノム解析
by
Haruka Ozaki
PDF
Up!winter2020
by
ssuserec0681
PPTX
2014年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
PPTX
ISMB2014読み会 Ragout—a reference-assisted assembly tool for bacterial genomes
by
Haruka Ozaki
フリーソフトではじめるがん体細胞変異解析入門 第33回勉強会資料
by
Amelieff
NGS現場の会第2回_アメリエフ株式会社_がんExome解析
by
Amelieff
Exome解析入門
by
Amelieff
フリーソフトではじめるNGS融合遺伝子解析入門
by
Amelieff
2014年度 第5回バイオインフォマティクス実習
by
Jun Nakabayashi
CBI学会2013チュートリアル NGSデータ解析入門 (解析編)配布資料
by
Genaris Omics, Inc.
RNASkim
by
弘毅 露崎
XCMSを用いた質量分析データ処理_BioCAsia2021_yamamoto.pdf
by
h_yama2396
2019年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
BGI Webinar Aug 28, 2014 "Genome wide methylation analysis and analytics"
by
kazuoishii20
NGS現場の会第2回_アメリエフ株式会社_RNAseq解析
by
Amelieff
2016年度 第4回バイオインフォマティクス実習
by
Jun Nakabayashi
20160324自由集会講演
by
astanabe
新規医療開発に関わる統計学 (バイオインフォマティクス)
by
Hidemasa Bono
2018年度 第1回バイオインフォマティクス実習
by
Jun Nakabayashi
2017年度 第1回バイオインフォマティクス実習
by
Jun Nakabayashi
Kashiwa.R#9 Rでゲノム解析
by
Haruka Ozaki
Up!winter2020
by
ssuserec0681
2014年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
ISMB2014読み会 Ragout—a reference-assisted assembly tool for bacterial genomes
by
Haruka Ozaki
フリーソフトではじめるChIP-seq解析_第40回勉強会資料
1.
フ リ ー
ソ フ ト で は じ め る C h I P - s e q 解 析 第 4 0 回 勉 強 会 資 料 2014年12月18日
2.
上記はほんの一部 日々、多くのソフトが公開されている Copyright © Amelieff
Corporation. All Rights Reserved. 2 N G S デ ー タ 解 析 の フ リ ー ソ フ ト QC • cutadapt • FastQC • FastX-toolkit • Trimmomaic : 多くのツールは公開されているフリーソフトであり、LinuxというOSで動作する アライメント • bowtie • bwa • BSMAP • Tophat : ピーク検出 ・アノテーション • MACS • QuEST • ChIPpeakAnno : ※Rなど、WindowsやMacでも動くものもある メチル化解析・比較 ・アノテーション • BSMAP • methylKit • BisSNP :
3.
Copyright © Amelieff
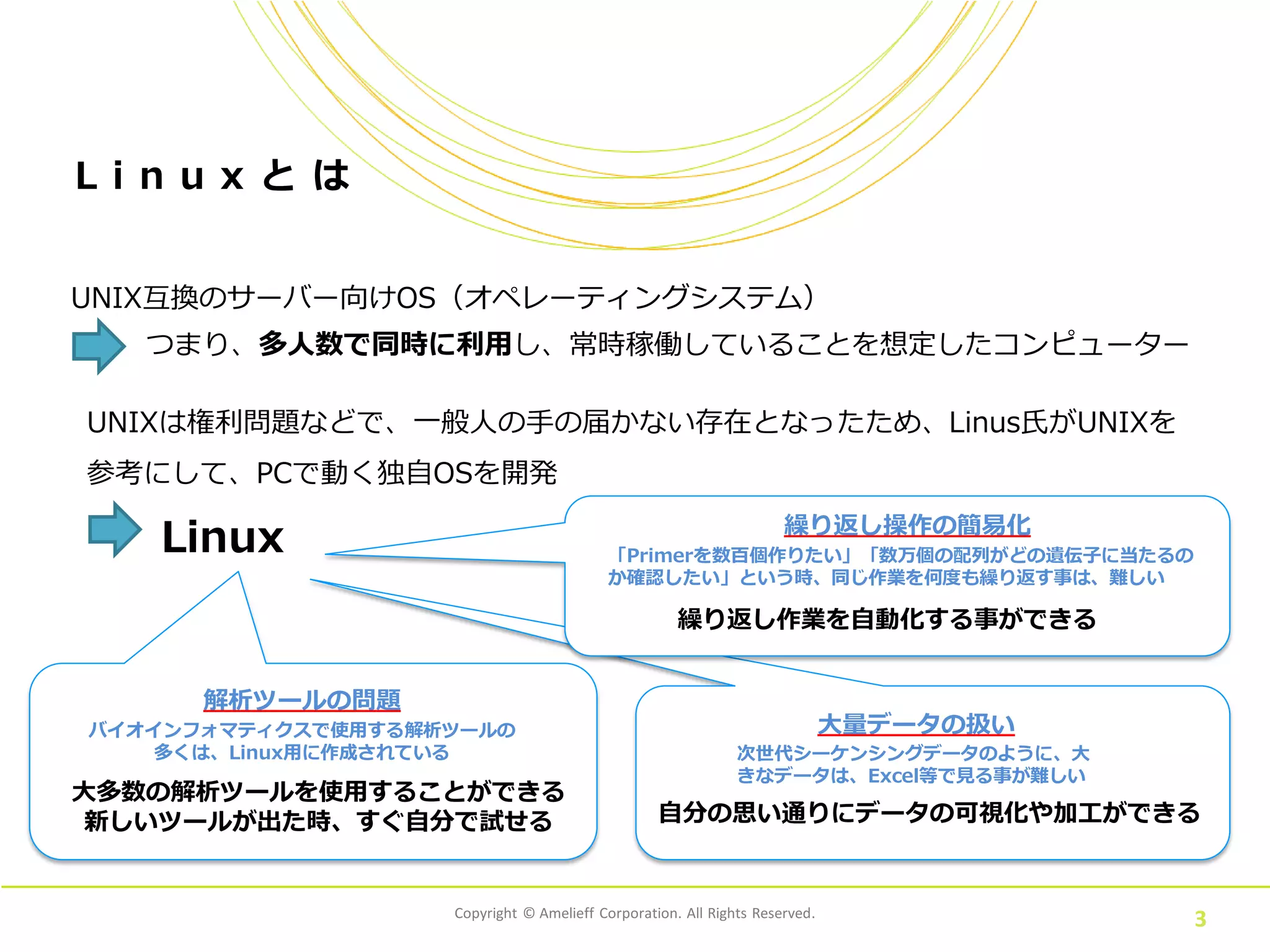
Corporation. All Rights Reserved. 3 L i n u x と は UNIX互換のサーバー向けOS(オペレーティングシステム) つまり、多人数で同時に利用し、常時稼働していることを想定したコンピューター UNIXは権利問題などで、一般人の手の届かない存在となったため、Linus氏がUNIXを 参考にして、PCで動く独自OSを開発 Linux 大多数の解析ツールを使用することができる 新しいツールが出た時、すぐ自分で試せる 次世代シーケンシングデータのように、大 きなデータは、Excel等で見る事が難しい 自分の思い通りにデータの可視化や加工ができる バイオインフォマティクスで使用する解析ツールの 多くは、Linux用に作成されている 「Primerを数百個作りたい」「数万個の配列がどの遺伝子に当たるの か確認したい」という時、同じ作業を何度も繰り返す事は、難しい 繰り返し作業を自動化する事ができる 大量データの扱い 繰り返し操作の簡易化 解析ツールの問題
4.
Copyright © Amelieff
Corporation. All Rights Reserved. 4 L i n u x と は Linuxにはさまざまなディストリビューション(配布形式)がある Debian系・・・Ubuntuなど Red Hat系・・・Red Hat Enterprise Linux(商用)、CentOS(無償)など 見た目やパッケージ管理形式が異なるが、基本的な操作コマンドは同じ 解析サーバにCentOSをお奨めする理由 • 更新方針が保守的で、アップデートが頻発しない • 枯れた技術を使っていて、安定している 弊社販売の 解析サーバで 使用
5.
Copyright © Amelieff
Corporation. All Rights Reserved. 5 解 析 手 法 の ご 紹 介 • 今回の解析で用いたサーバ OS CentOS6 64bit CPU Intel Corei7-3930K[3.2GHz/6Core] メモリ 64GB SSD 64GB(OS用) HDD 2TB × 4台 時間がかかる処理については実行時間を示します
6.
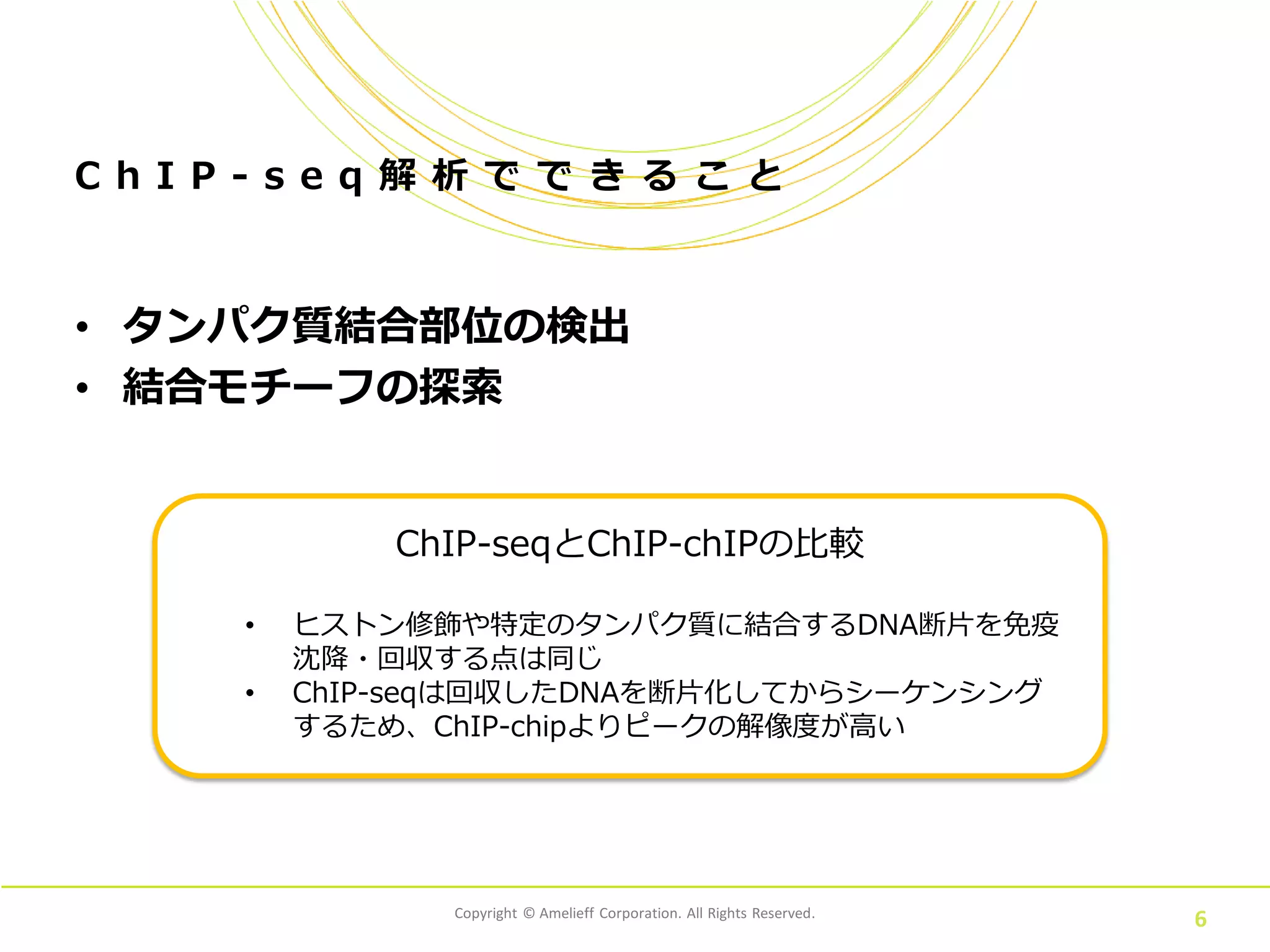
Copyright © Amelieff
Corporation. All Rights Reserved. C h I P - s e q 解 析 で で き る こ と • タンパク質結合部位の検出 • 結合モチーフの探索 6 ChIP-seqとChIP-chIPの比較 • ヒストン修飾や特定のタンパク質に結合するDNA断片を免疫 沈降・回収する点は同じ • ChIP-seqは回収したDNAを断片化してからシーケンシング するため、ChIP-chipよりピークの解像度が高い
7.
Copyright © Amelieff
Corporation. All Rights Reserved. 7 用 い た テ ス ト デ ー タ • NCBI GEOに登録されているヒトのChIP-seqデータ – GSM1295084: BF細胞(ヒト成人繊維芽細胞)のH3K27me3 ChIP-seq • SRA ID:SRR1055695 – GSM1295086: BF細胞のH3 input(コントロール) ChipSeq • SRA ID:SRR1055697 – すべてIllumina GAIIx、36bp Single-End – URL:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40740 H3K27me3 • ヒストンH3の27番目のリジンのトリメチル化→転写抑制に関与
8.
Copyright © Amelieff
Corporation. All Rights Reserved. 8 解 析 手 法 の ご 紹 介 • GEOからダウンロードしたファイルはSRAフォーマットという独自形式に なっており、そのままでは解析に使えない • NCBI SRA Toolkitを使ってSRAフォーマットをFASTQフォーマットに変換 クオリティコントロール → マッピング→ピーク検出→アノテーション $ fastq-dump SRR1055695.sra $ fastq-dump SRR1055697.sra 拡張子が「.fastq」のFASTQファイルができる • データのクオリティを集計して可視化する $ fastqc -o 1_qc -f fastq SRR1055695.fastq $ fastqc -o 1_qc -f fastq SRR1055697.fastq
9.
Copyright © Amelieff
Corporation. All Rights Reserved. 9 解 析 手 法 の ご 紹 介 • クオリティの低い塩基・リードを除去する $ fastq_quality_trimmer -t 20 -l 30 -Q 33 -i SRR1055695.fastq | fastq_quality_filter -q 20 -p 80 -Q 33 -o 1_qc/SRR1055695.clean.fastq 3’末端からクオリティ20未満の塩基をトリミングし、長さが30塩基未満になった リードを破棄する 80%以上の塩基がクオリティー20以上のリードのみを抽出する 約3分 他のFASTQファイルに対しても実施する クオリティコントロール → マッピング→ピーク検出→アノテーション
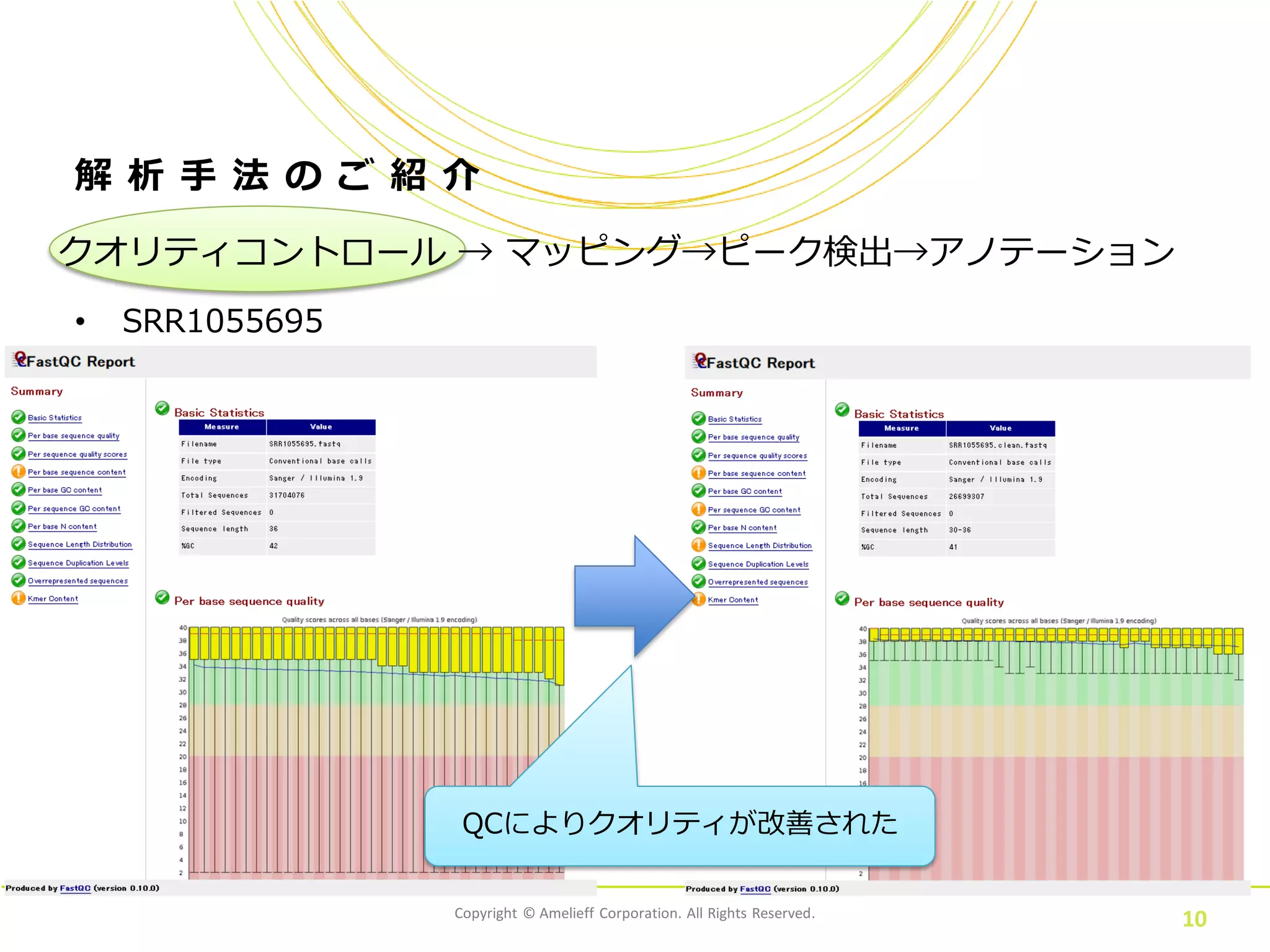
10.
Copyright © Amelieff
Corporation. All Rights Reserved. 10 解 析 手 法 の ご 紹 介 • SRR1055695 クオリティコントロール → マッピング→ピーク検出→アノテーション QCによりクオリティが改善された
11.
Copyright © Amelieff
Corporation. All Rights Reserved. 11 解 析 手 法 の ご 紹 介 • SRR1055697 クオリティコントロール → マッピング→ピーク検出→アノテーション QCによりクオリティが改善された
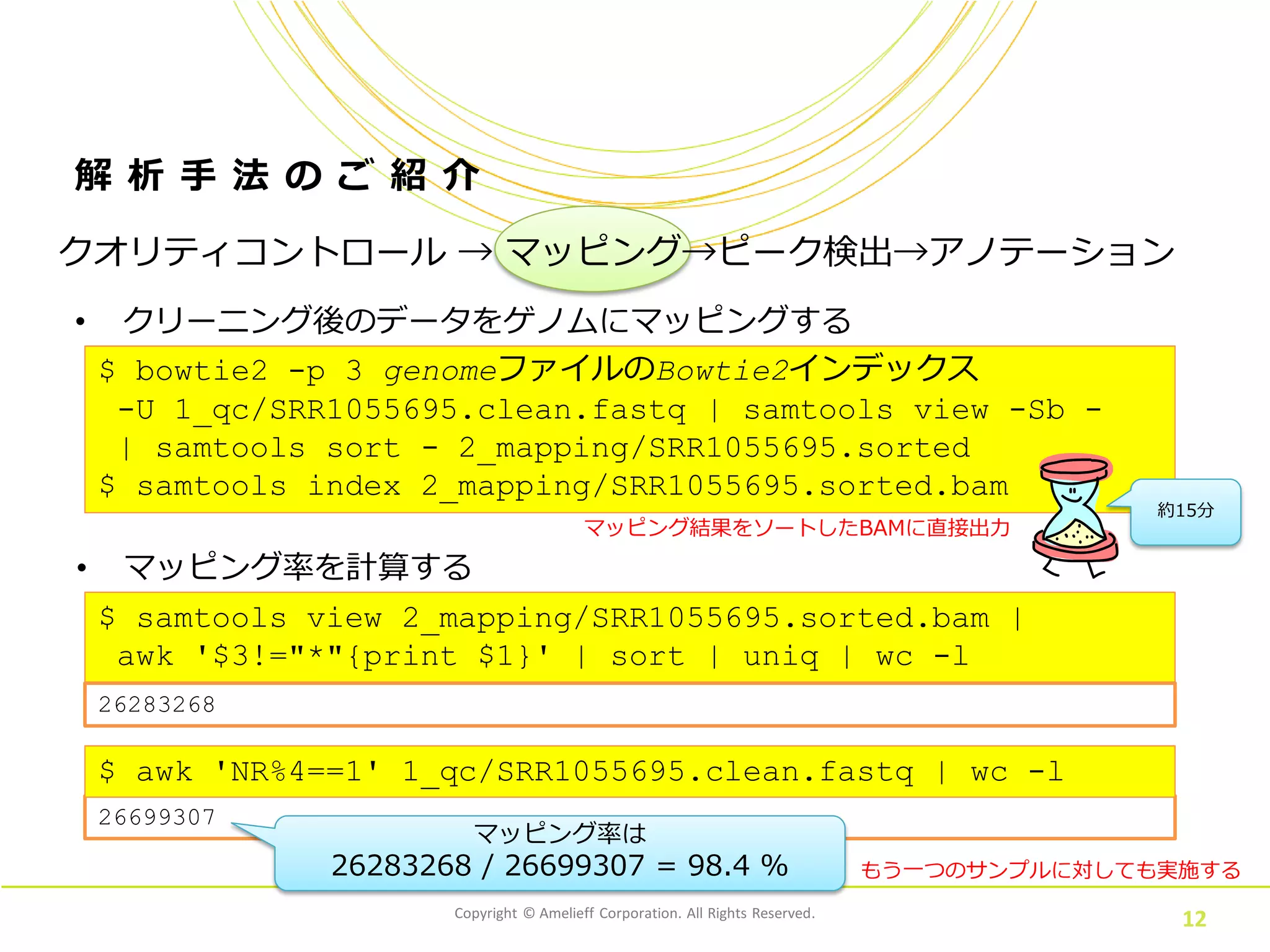
12.
$ samtools view
2_mapping/SRR1055695.sorted.bam | awk '$3!="*"{print $1}' | sort | uniq | wc -l Copyright © Amelieff Corporation. All Rights Reserved. 12 解 析 手 法 の ご 紹 介 • クリーニング後のデータをゲノムにマッピングする $ bowtie2 -p 3 genomeファイルのBowtie2インデックス -U 1_qc/SRR1055695.clean.fastq | samtools view -Sb - | samtools sort - 2_mapping/SRR1055695.sorted $ samtools index 2_mapping/SRR1055695.sorted.bam 約15分 もう一つのサンプルに対しても実施する クオリティコントロール → マッピング→ピーク検出→アノテーション • マッピング率を計算する 他のサンプルに対しても実施する26699307 マッピング率は 26283268 / 26699307 = 98.4 % $ awk 'NR%4==1' 1_qc/SRR1055695.clean.fastq | wc -l 26283268 マッピング結果をソートしたBAMに直接出力
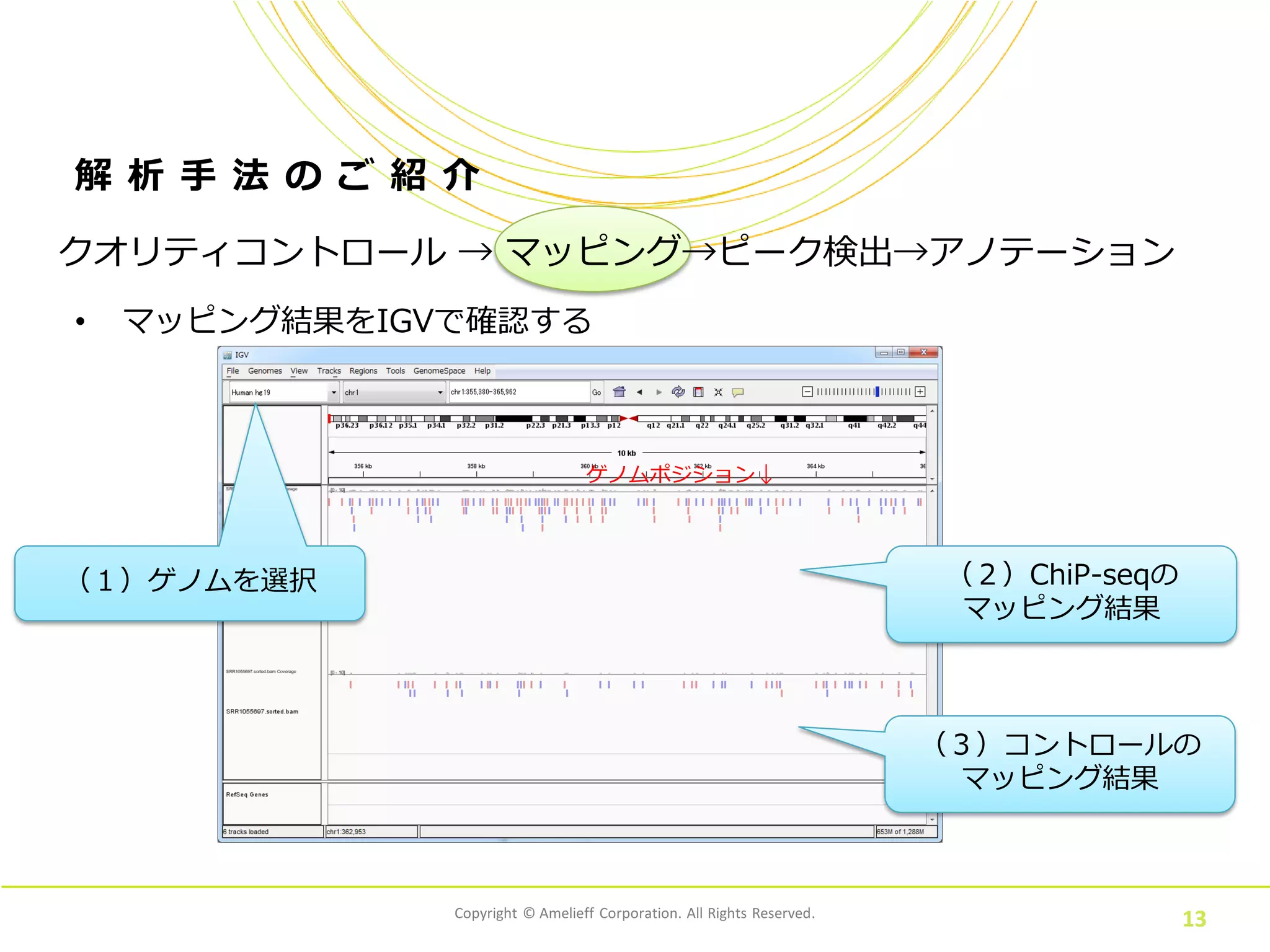
13.
Copyright © Amelieff
Corporation. All Rights Reserved. 13 解 析 手 法 の ご 紹 介 • マッピング結果をIGVで確認する クオリティコントロール → マッピング→ピーク検出→アノテーション (1)ゲノムを選択 (2)ChiP-seqの マッピング結果 ゲノムポジション↓ (3)コントロールの マッピング結果
14.
Copyright © Amelieff
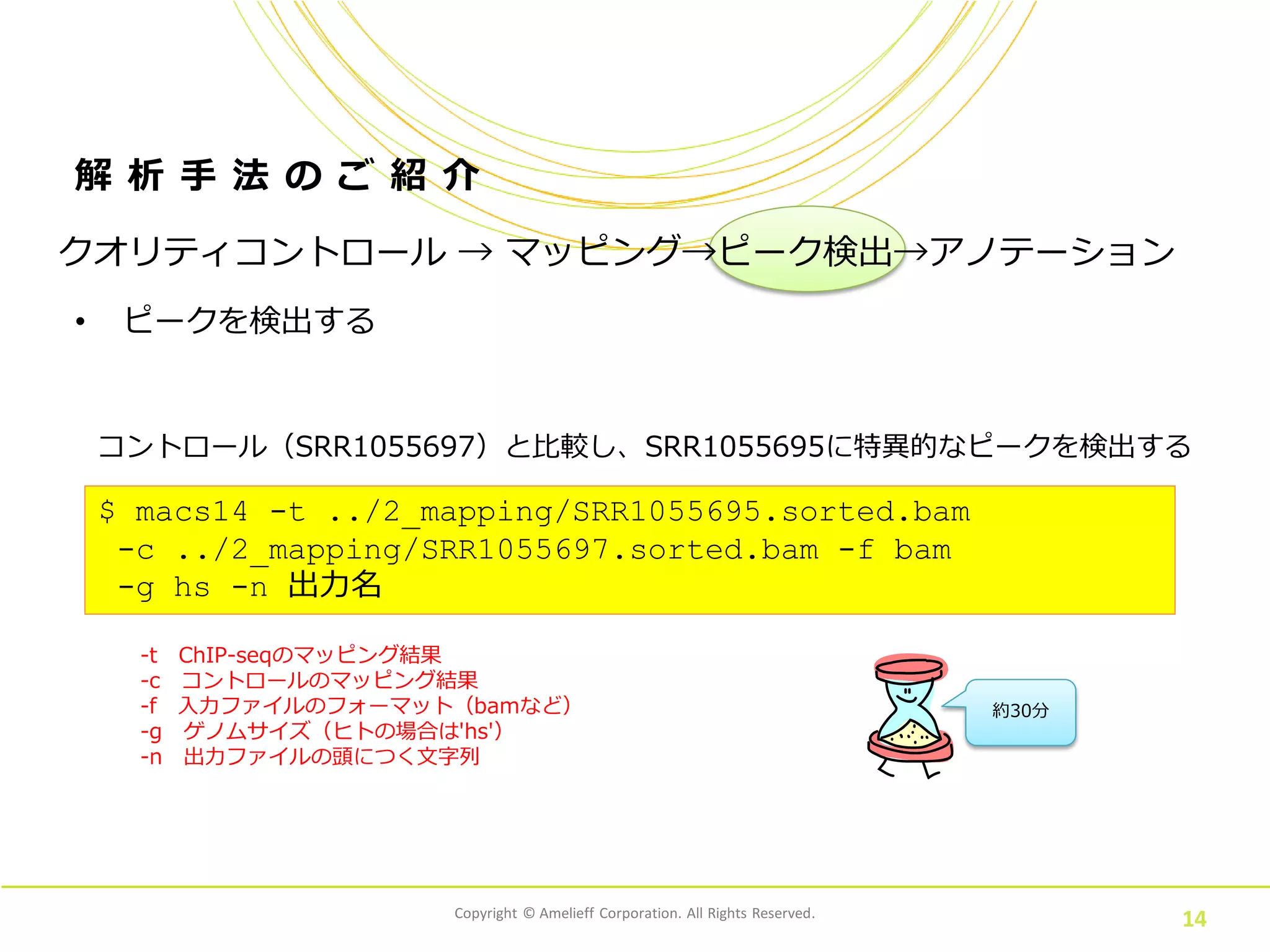
Corporation. All Rights Reserved. 14 解 析 手 法 の ご 紹 介 • ピークを検出する $ macs14 -t ../2_mapping/SRR1055695.sorted.bam -c ../2_mapping/SRR1055697.sorted.bam -f bam -g hs -n 出力名 コントロール(SRR1055697)と比較し、SRR1055695に特異的なピークを検出する 約30分 クオリティコントロール → マッピング→ピーク検出→アノテーション -t ChIP-seqのマッピング結果 -c コントロールのマッピング結果 -f 入力ファイルのフォーマット(bamなど) -g ゲノムサイズ(ヒトの場合は'hs') -n 出力ファイルの頭につく文字列
15.
Copyright © Amelieff
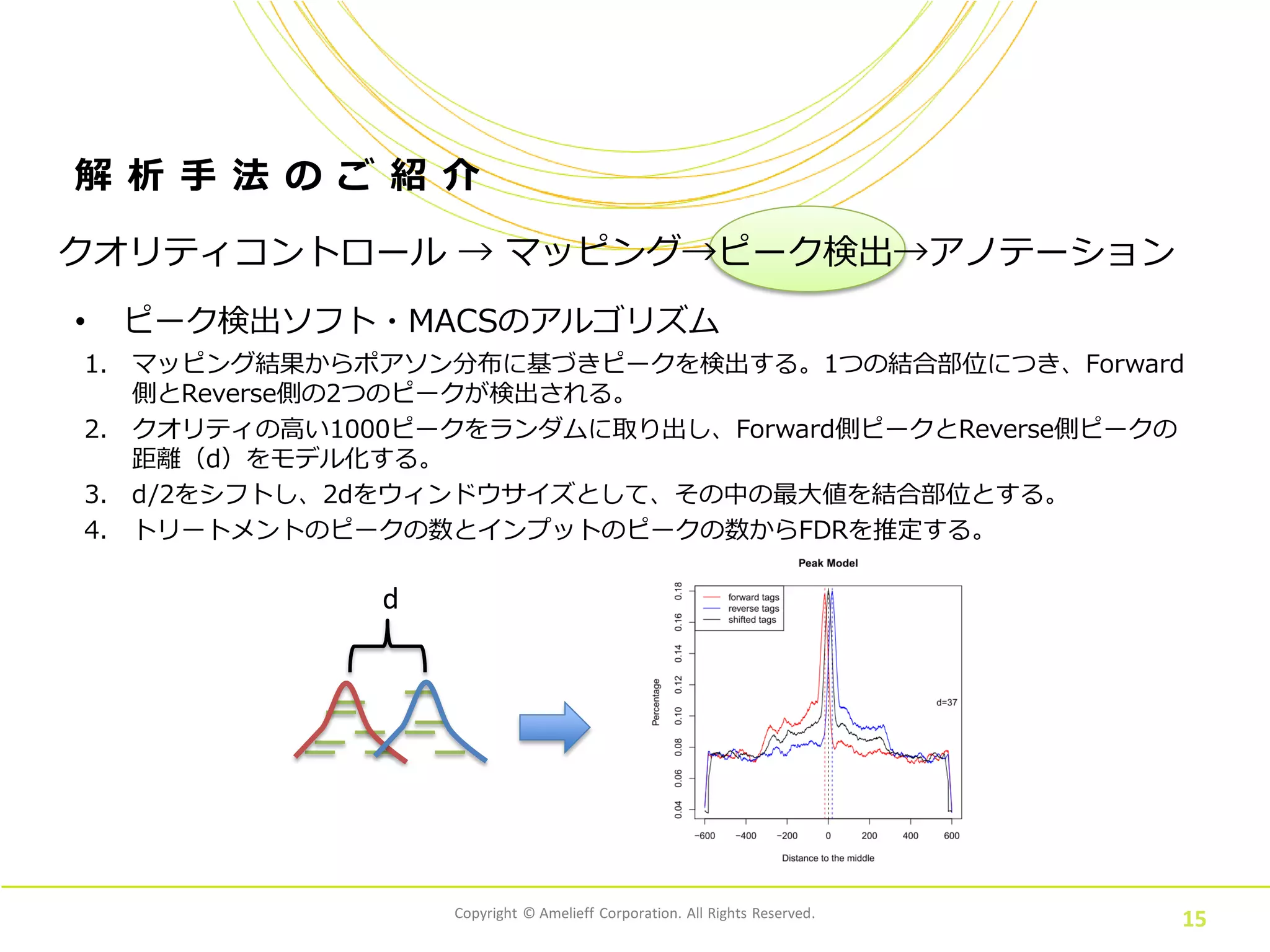
Corporation. All Rights Reserved. 15 解 析 手 法 の ご 紹 介 • ピーク検出ソフト・MACSのアルゴリズム クオリティコントロール → マッピング→ピーク検出→アノテーション 1. マッピング結果からポアソン分布に基づきピークを検出する。1つの結合部位につき、Forward 側とReverse側の2つのピークが検出される。 2. クオリティの高い1000ピークをランダムに取り出し、Forward側ピークとReverse側ピークの 距離(d)をモデル化する。 3. d/2をシフトし、2dをウィンドウサイズとして、その中の最大値を結合部位とする。 4. トリートメントのピークの数とインプットのピークの数からFDRを推定する。 d
16.
Copyright © Amelieff
Corporation. All Rights Reserved. 16 解 析 手 法 の ご 紹 介 • ピーク検出結果(xx_peaks.bed) クオリティコントロール → マッピング→ピーク検出→アノテーション 1. 染色体名 2. ピーク開始ポジション(0スタート) 3. ピーク終了ポジション 4. ピーク名 5. スコア:ピークの -10*log10(pvalue)
17.
Copyright © Amelieff
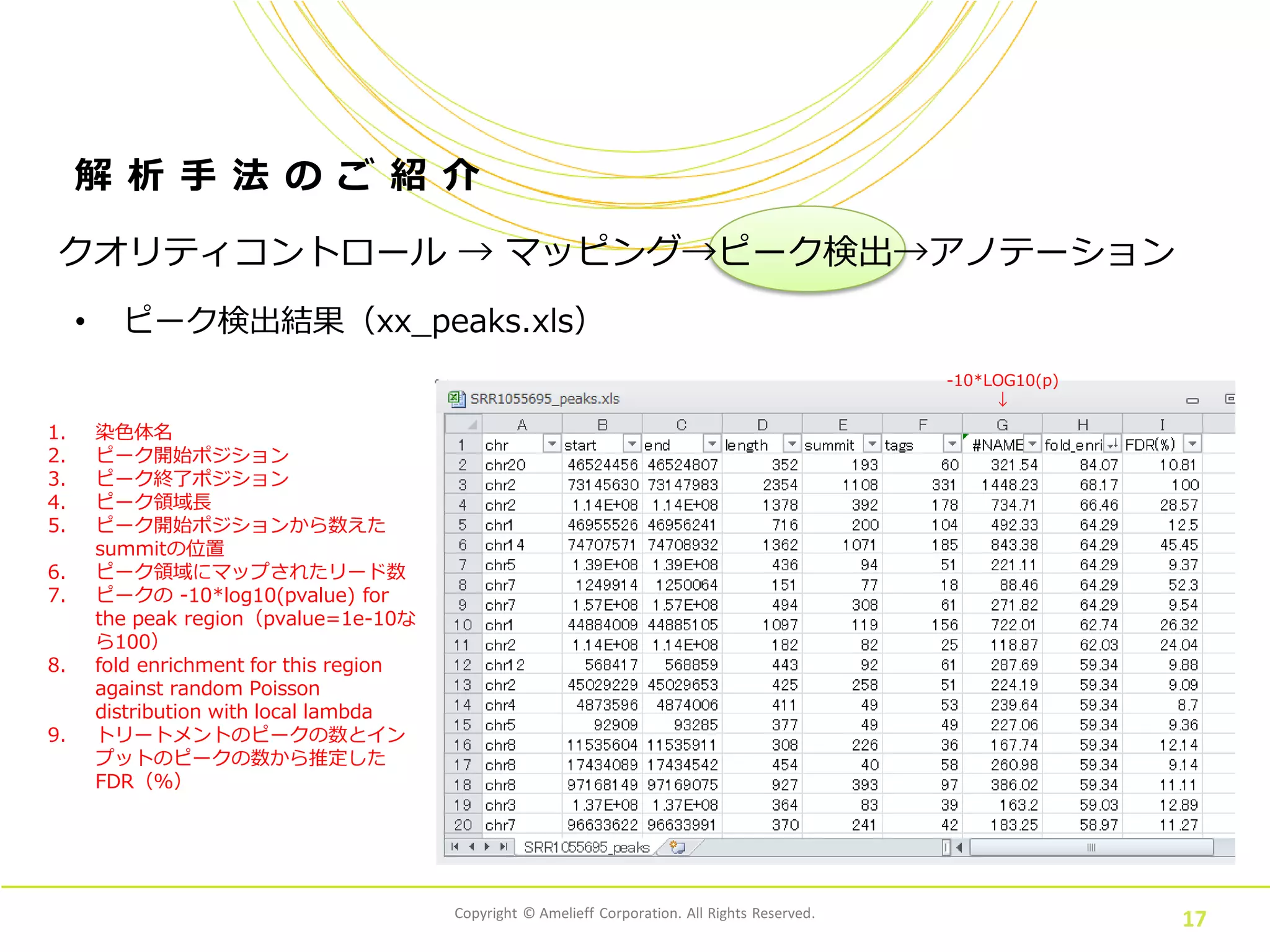
Corporation. All Rights Reserved. 17 解 析 手 法 の ご 紹 介 • ピーク検出結果(xx_peaks.xls) クオリティコントロール → マッピング→ピーク検出→アノテーション 1. 染色体名 2. ピーク開始ポジション 3. ピーク終了ポジション 4. ピーク領域長 5. ピーク開始ポジションから数えた summitの位置 6. ピーク領域にマップされたリード数 7. ピークの -10*log10(pvalue) for the peak region(pvalue=1e-10な ら100) 8. fold enrichment for this region against random Poisson distribution with local lambda 9. トリートメントのピークの数とイン プットのピークの数から推定した FDR(%) -10*LOG10(p) ↓
18.
Copyright © Amelieff
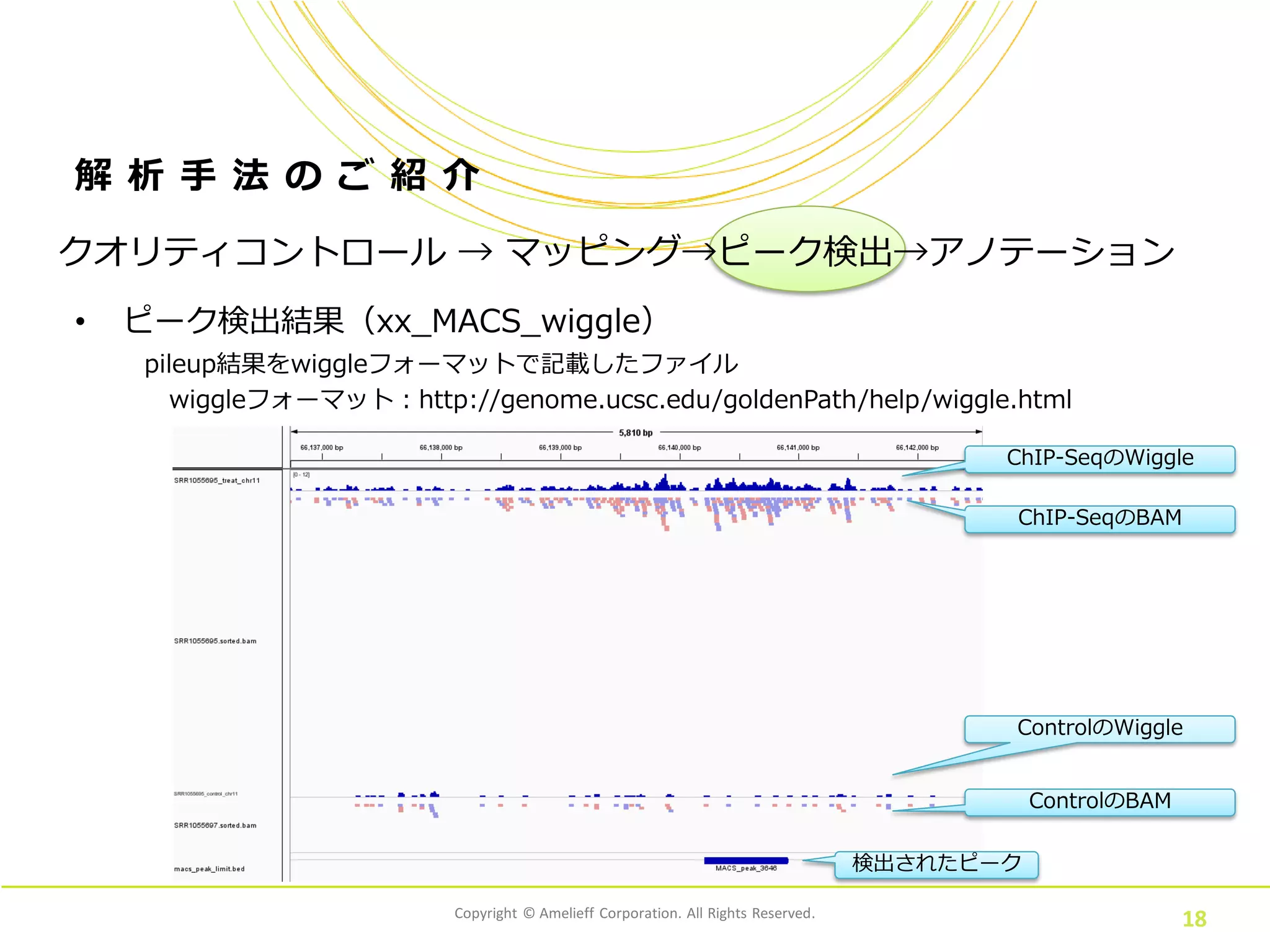
Corporation. All Rights Reserved. 18 解 析 手 法 の ご 紹 介 • ピーク検出結果(xx_MACS_wiggle) クオリティコントロール → マッピング→ピーク検出→アノテーション pileup結果をwiggleフォーマットで記載したファイル wiggleフォーマット:http://genome.ucsc.edu/goldenPath/help/wiggle.html ChIP-SeqのWiggle 検出されたピーク ChIP-SeqのBAM ControlのWiggle ControlのBAM
19.
18,520 429 76
0 Copyright © Amelieff Corporation. All Rights Reserved. 19 解 析 手 法 の ご 紹 介 • FDRの小さいものに絞り込む クオリティコントロール → マッピング→ピーク検出→アノテーション 全結果 FDR<10% FDR<9% FDR<8% [1] Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. Epub 2008 Sep 17. PubMed PMID: 18798982; PubMed Central PMCID: PMC2592715. MACSの論文[1]ではFDR<1%に絞っていたが 今回のデータはFDRが大きかったため FDR<9%に絞った
20.
Copyright © Amelieff
Corporation. All Rights Reserved. 20 ピ ー ク 検 出 ソ フ ト Q u E S T • 他の検出ソフト(QuEST)でも実行 – QuESTの特徴:実行時に結合タンパクの種類を選べる – 入力フォーマットはMAQ、ELAND、bowtieなど(BAMには未対応) $ bowtie -p 3 genomeファイルのBowtieインデックス 1_qc/SRR1055695.clean.fastq 2_mapping/SRR1055695.bowtie bowtieによるマッピング(bowtieフォーマットで出力) $ bowtie -p 3 genomeファイルのBowtieインデックス 1_qc/SRR1055697.clean.fastq 2_mapping/SRR1055697.bowtie 各約5分
21.
Copyright © Amelieff
Corporation. All Rights Reserved. 21 ピ ー ク 検 出 ソ フ ト Q u E S T • 他の検出ソフト(QuEST)でも実行 SRR1055695.3 YILLUMINA-B8EC94_105:4:1:1555:1140 length=36 - chr22 32873017 ACACATAGTTCATTTGAGGTGTTTTTGCTTTTTCTG FGDGEDGEFFGGGEGEGGDD>@HHHHHHHFHGEHHH 0 SRR1055695.4 YILLUMINA-B8EC94_105:4:1:1583:1139 length=36 - chr12 34846311 TGAAACACTCTGTTTGTAAAGTCTGCACGTGGATAT DGHGHHHHHHHHHHHHHHHHHGBGBEHHHHHHFHGH 0 SRR1055695.2 YILLUMINA-B8EC94_105:4:1:1226:1131 length=36 + chr12 5193061 TTTTCTCTTATCTTTTCTAAAANTCNTAAACTAGGT GGGG8EDGGDEDGGGDDDDD=:#;;#;:9<BEEE@D 0 22:T>N,25:C>N : bowtieフォーマット
22.
Copyright © Amelieff
Corporation. All Rights Reserved. 22 ピ ー ク 検 出 ソ フ ト Q u E S T • 他の検出ソフト(QuEST)でも実行 QuESTを実行 $ generate_QuEST_parameters.pl -rp 染色体Fastaのディレクトリ/ -gt genome.fa.faiのパス -bowtie_align_ChIP SRR1055695.bowtie -bowtie_align_RX_noIP SRR1055697.bowtie -ap 出力ディレクトリ • -rp リファレンスゲノムの染色体ごとのFastaを置いたディレクトリ • -gt リファレンスゲノムの染色体名とサイズの組のリスト • -bowtie_align_ChIP ChIP-seqのマッピング結果(bowtieフォーマット) • -bowtie_align_RX_noIP Controlのマッピング結果(bowtieフォーマット) • -ap 結果出力ディレクトリ
23.
Copyright © Amelieff
Corporation. All Rights Reserved. 23 ピ ー ク 検 出 ソ フ ト Q u E S T • 他の検出ソフト(QuEST)でも実行 QuESTを実行 結合タンパクの種類 ・転写因子→1 ・ PolII-like factor→2 ・ヒストン→3 ・自分でパラメータを設定→4 「3」を選択
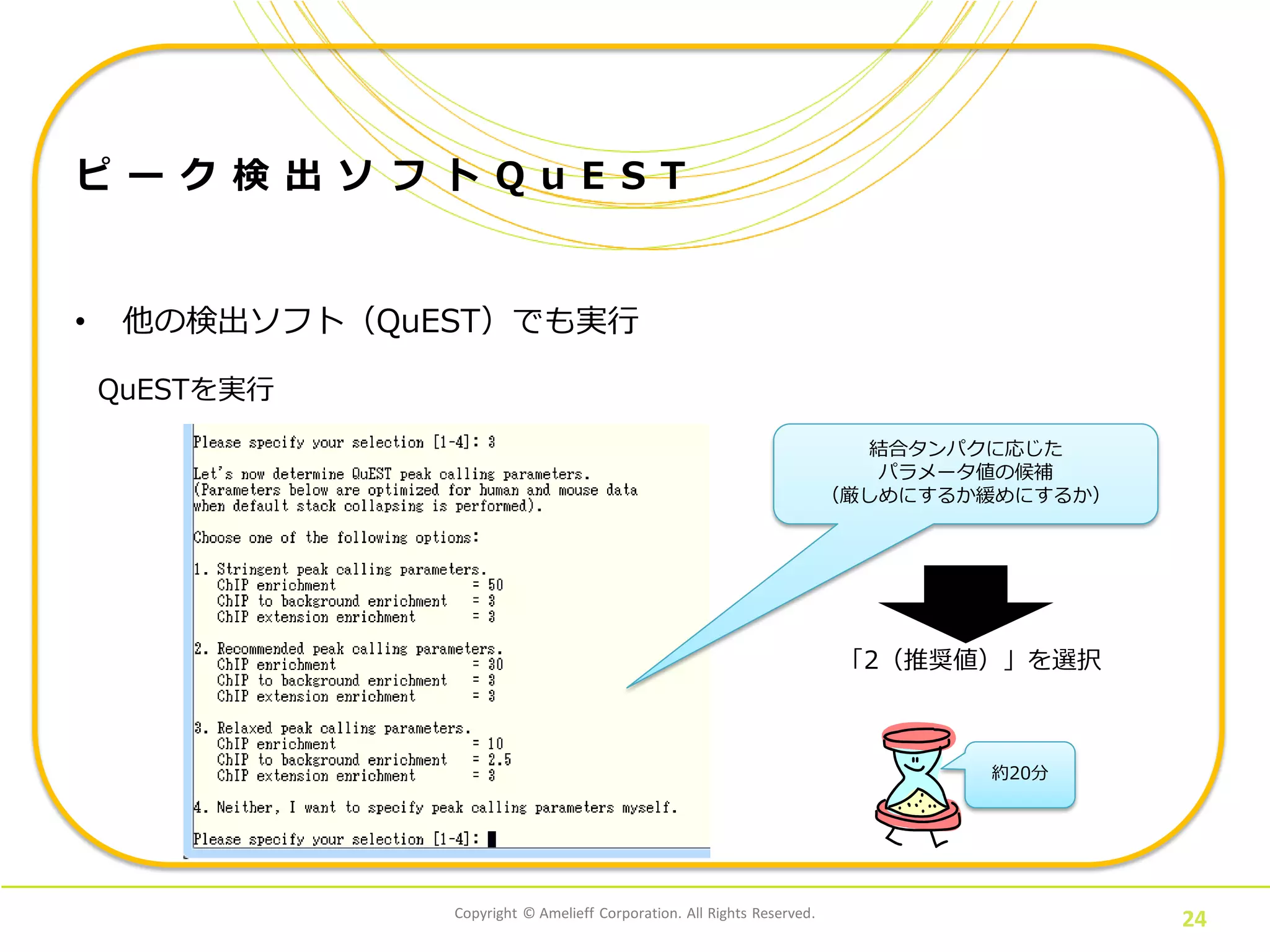
24.
Copyright © Amelieff
Corporation. All Rights Reserved. 24 ピ ー ク 検 出 ソ フ ト Q u E S T • 他の検出ソフト(QuEST)でも実行 QuESTを実行 結合タンパクに応じた パラメータ値の候補 (厳しめにするか緩めにするか) 「2(推奨値)」を選択 約20分
25.
Copyright © Amelieff
Corporation. All Rights Reserved. 25 ピ ー ク 検 出 ソ フ ト Q u E S T • QuESTの結果:概要(module_outputs/QuEST.out) bowtieフォーマット ## please cite: ## Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, ## Myers RM, Sidow A ## Genome-wide analysis of transcription factor binding sites based ## on ChIP-Seq data. ## Nat Methods. 2008 Sep; 5:(9):829-35 ChIP peaks: 13 ChIP peaks accepted: 13 ChIP peaks rejected: 0 ChIP regions: 11 ChIP regions accepted: 11 ChIP regions rejected: 0
26.
Copyright © Amelieff
Corporation. All Rights Reserved. 26 ピ ー ク 検 出 ソ フ ト Q u E S T • QuESTの結果:ピーク(calls/peak_caller.ChIP.out.accepted) bowtieフォーマット R-1 chr11 3674740-3676339 ChIP: 170.5 control: 22.6886 max_pos: 3675711 ef: 7.51476 ChIP_tags: 1572 background_tags: 118 tag_ef: 7.92329 ps: 17 cor: 0.485103 -log10_qv: 29914.9 -log10_pv: 29922.1 qv_rank: 1 P-1-1 chr11 3675169 ChIP: 123.088 control: 12.954 region: 3674740-3676340 ef: 9.50194 ps: 17 cor: 0.980156 -log10_qv: 231.8 -log10_pv: 238.99 qv_rank: 5 P-1-2 chr11 3675711 ChIP: 170.5 control: 22.6886 region: 3674740-3676340 ef: 7.51476 ps: 15 cor: 0.94687 -log10_qv: 238.543 -log10_pv: 245.732 qv_rank: 4 R-2 chr22 51081718-51084682 ChIP: 48.6881 control: 3.90919 max_pos: 51082255 ef: 12.4548 ChIP_tags: 430 background_tags: 22 tag_ef: 11.6247 ps: 13 cor: 0.171511 -log10_qv: 1053.29 - log10_pv: 1059.78 qv_rank: 5 P-2-1 chr22 51082255 ChIP: 48.6881 control: 3.90919 region: 51081718-51084683 ef: 12.4548 ps: 13 cor: 0.875156 -log10_qv: 571.506 -log10_pv: 577.997 qv_rank: 1 : ピークの位置、スコア、q-valueなどが記載されている
27.
Copyright © Amelieff
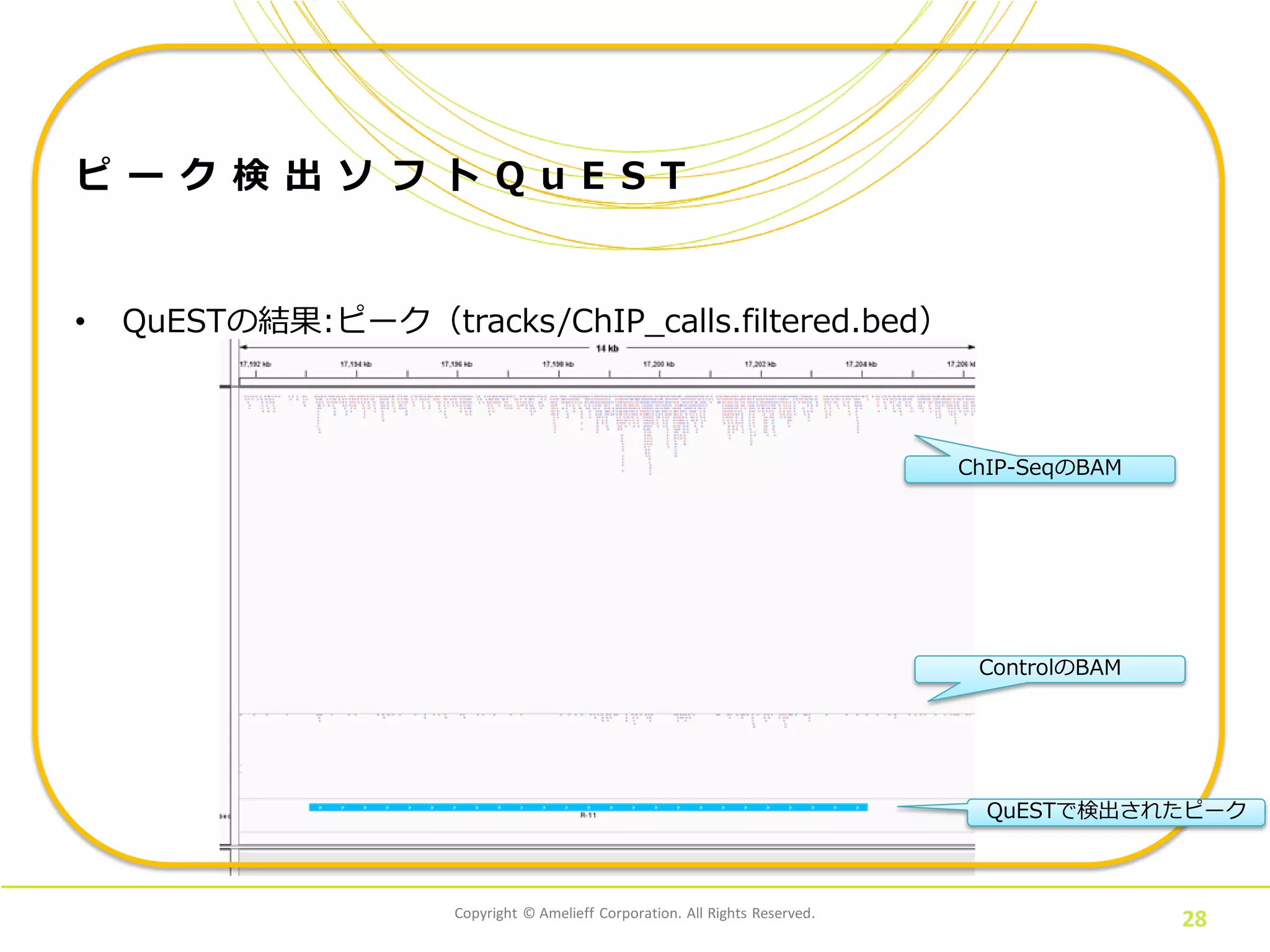
Corporation. All Rights Reserved. 27 ピ ー ク 検 出 ソ フ ト Q u E S T • QuESTの結果:ピーク(tracks/ChIP_calls.filtered.bed) track name=ChIP_filtered description=ChIP_filtered_regions itemRgb="On" priority=67 visibility=1 chr11 3674741 3676340 R-1 170.5 + 3674741 3676340 0,191,255 chr22 51081719 51084683 R-2 48.6881 + 51081719 51084683 0,191,255 chr17 153120 155470 R-3 43.6734 + 153120 155470 0,191,255 chr7 100547703 100553968 R-4 42.4881 + 100547703 100553968 0,191,255 chr20 46522428 46525209 R-5 35.9235 + 46522428 46525209 0,191,255 chr18 111978 112686 R-6 35.4676 + 111978 112686 0,191,255 chr20 62719779 62720414 R-7 34.2823 + 62719779 62720414 0,191,255 chr2 133021646 133031873 R-8 33.4617 + 133021646 133031873 0,191,255 chr7 944472 946396 R-9 32.6411 + 944472 946396 0,191,255 chr2 90448094 90454767 R-10 32.5499 + 90448094 90454767 0,191,255 chr1 17193078 17204129 R-11 30.6352 + 17193078 17204129 0,191,255 : IGVで表示するために以下の処理が必要 ・ピークとSummitの情報が混在しているので、分ける ・track行を除去しておく
28.
Copyright © Amelieff
Corporation. All Rights Reserved. 28 ピ ー ク 検 出 ソ フ ト Q u E S T • QuESTの結果:ピーク(tracks/ChIP_calls.filtered.bed) ChIP-SeqのBAM ControlのBAM QuESTで検出されたピーク
29.
Copyright © Amelieff
Corporation. All Rights Reserved. 29 ピ ー ク 検 出 ソ フ ト Q u E S T • MACSとQuESTで共通するピークを探す $ intersectBed -wa -a macsの.bed -b QuESTのbed MACSのピークのうち、QuESTのピークとオーバーラップするものを探す $ intersectBed -v -a macsの.bed -b QuESTのbed MACSのピークのうち、QuESTのピークとオーバーラップしないものを探す
30.
Copyright © Amelieff
Corporation. All Rights Reserved. 30 解 析 手 法 の ご 紹 介 • ピークをアノテーション クオリティコントロール → マッピング→ピーク検出→アノテーション MACSで検出されたピークの読み込みと変換 > macs_bed = read.table("MACSのbed") > macs = BED2RangedData(macs_bed) Rを起動し、ChIPpeakAnnoパッケージを読み込む $ R > library(ChIPpeakAnno) EBI Biomartからヒト遺伝子情報をダウンロードし、アノテーション > mart = useMart(biomart="ensembl", dataset="hsapiens_gene_ensembl") > myAnno = getAnnotation(mart) > annotatedPeak = annotatePeakInBatch(macs, AnnotationData=myAnno)
31.
Copyright © Amelieff
Corporation. All Rights Reserved. 31 解 析 手 法 の ご 紹 介 • ピークをアノテーション クオリティコントロール → マッピング→ピーク検出→アノテーション アノテーション結果をファイル出力 > write.table(as.data.frame(annotatedPeak), file="annotatedPeakList.tsv", sep="¥t", row.names=FALSE) 近傍遺伝子からの距離
32.
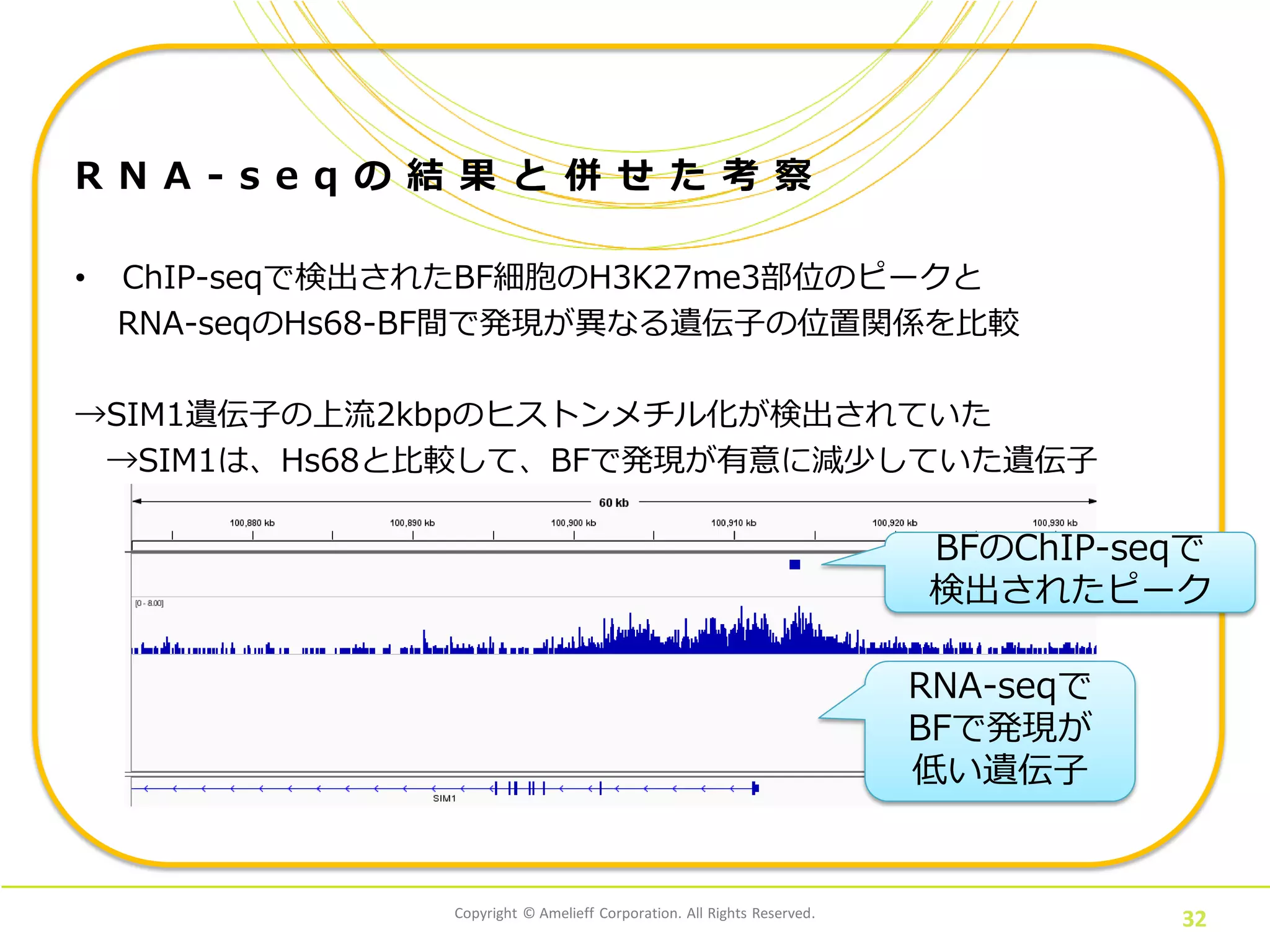
Copyright © Amelieff
Corporation. All Rights Reserved. 32 R N A - s e q の 結 果 と 併 せ た 考 察 • ChIP-seqで検出されたBF細胞のH3K27me3部位のピークと RNA-seqのHs68-BF間で発現が異なる遺伝子の位置関係を比較 →SIM1遺伝子の上流2kbpのヒストンメチル化が検出されていた →SIM1は、Hs68と比較して、BFで発現が有意に減少していた遺伝子 BFのChIP-seqで 検出されたピーク RNA-seqで BFで発現が 低い遺伝子
33.
Copyright © Amelieff
Corporation. All Rights Reserved. 33 R N A - s e q の 結 果 と 併 せ た 考 察 • SIM1は胎児の腎臓で特異的に発現することが報告されている • メチル化と遺伝子発現の関係を検証するには、さらに以下のような解析が必要 (※本日は紹介しない) → Hs68のChIP-seq結果との比較 → パスウェイ解析、Gene Ontology解析 など 成人由来BFで発現が低下している のと関連している可能性がある
34.
Copyright © Amelieff
Corporation. All Rights Reserved. 34 共 通 モ チ ー フ 検 索 • MEME(http://meme.nbcr.net/meme/intro.html)などで、ピークに 共通するモチーフ配列を検索 ピーク領域の塩基配列を取得 $ samtools faidx genomeファイルのFasta chr1:17193078-17204129 $ samtools faidx genomeファイルのFasta chr2:90446939-90454767 : (各ピークに対して実行) 上記の結果からFastaファイルを作成
35.
Copyright © Amelieff
Corporation. All Rights Reserved. 35 共 通 モ チ ー フ 検 索 • MEME(http://meme.nbcr.net/meme/intro.html)などで、ピークに 共通するモチーフ配列を検索
36.
Copyright © Amelieff
Corporation All Rights Reserved. 36 アメリエフ バイオインフォマティクス 調査リクエストサービス バイオ研究の解析に使用するソフトや解析手法について、 無償で調査するサービスです。調査結果はアメリエフの ブログでご紹介いたします。 申込みフォーム http://goo.gl/g3SOtU ア メ リ ク



![Copyright © Amelieff Corporation. All Rights Reserved.
5
解 析 手 法 の ご 紹 介
• 今回の解析で用いたサーバ
OS CentOS6 64bit
CPU Intel Corei7-3930K[3.2GHz/6Core]
メモリ 64GB
SSD 64GB(OS用)
HDD 2TB × 4台
時間がかかる処理については実行時間を示します](https://image.slidesharecdn.com/40chip-seqslideshare-141218233848-conversion-gate02/75/ChIP-seq-_-40-5-2048.jpg)





![18,520 429 76 0
Copyright © Amelieff Corporation. All Rights Reserved.
19
解 析 手 法 の ご 紹 介
• FDRの小さいものに絞り込む
クオリティコントロール → マッピング→ピーク検出→アノテーション
全結果 FDR<10% FDR<9% FDR<8%
[1] Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS.
Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. Epub 2008 Sep 17.
PubMed PMID: 18798982; PubMed Central PMCID: PMC2592715.
MACSの論文[1]ではFDR<1%に絞っていたが
今回のデータはFDRが大きかったため
FDR<9%に絞った](https://image.slidesharecdn.com/40chip-seqslideshare-141218233848-conversion-gate02/75/ChIP-seq-_-40-19-2048.jpg)