1. The document discusses a contact map prediction method that was one of the top performers in CASP8. It uses topological properties of proteins like recursive convex hulls along with machine learning to predict contacts.
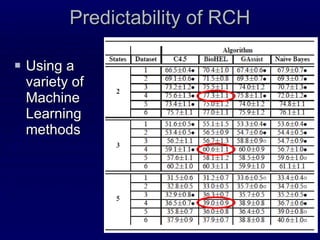
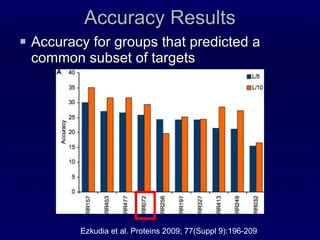
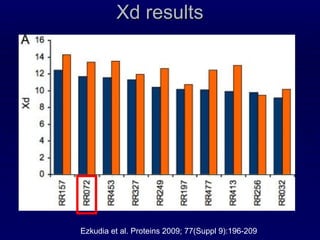
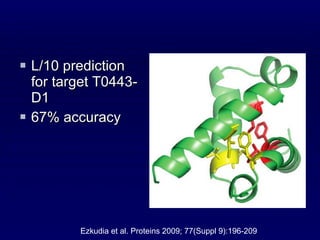
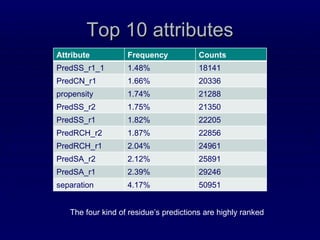
2. The method was tested on CASP8 targets, achieving high accuracy scores. Analysis of the predictive rules provided insights into which features were most important.
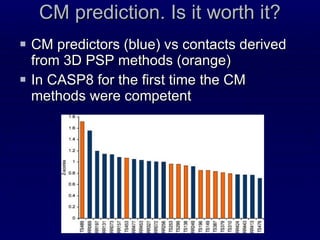
3. While performance was good, there is still room for improvement, such as better ranking of predictions or using correlated mutations. The contact map prediction approach is valuable as in CASP8 it was competitive with methods using 3D structure prediction.
![Data Mining Protein Structures' Topological Properties to Enhance Contact Map Predictions Dr. Jaume Bacardit School of Computer Science and School of Biosciences University of Nottingham [email_address] Weizmann Institute of Sciences, May 27 th , 2010](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-1-320.jpg)
![Data Mining Protein Structures' Topological Properties to Enhance Contact Map Predictions Dr. Jaume Bacardit School of Computer Science and School of Biosciences University of Nottingham [email_address] Weizmann Institute of Sciences, May 27 th , 2010](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/75/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-1-2048.jpg)







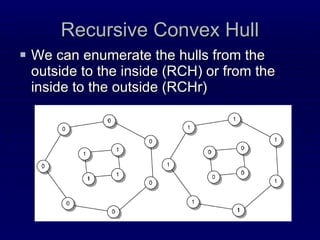
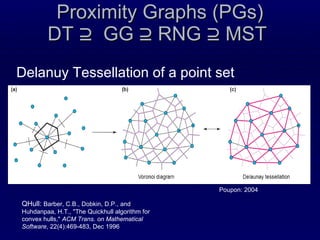
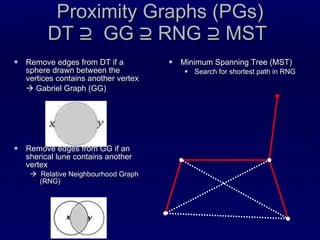
![Recursive Convex Hull Structural feature that we have proposed recently [Stout, Bacardit, Hirst & Krasnogor, Bioinformatics 2008 24(7):916-923; ] We model a protein as a series of nested layers, assigning each residue to a different layer Strictly speaking each layer is a convex hull of points The convex hull of a point set is simple and fast to compute Recursive Convex Hull is computed by iteratively identifying the layers (hulls) of a protein](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-11-320.jpg)
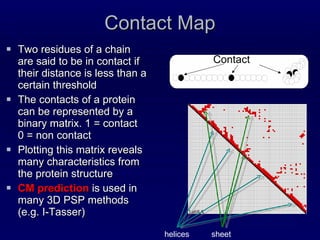
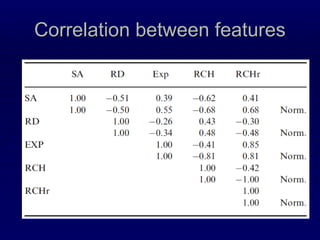
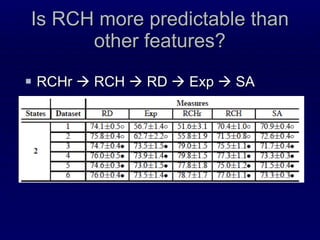
![Relation of RCH to other structural properties Comparing Solvent Accessiblity Exposure [Ben-Shimon and Eisenstein;05] Residue depth [Chakravarti and Varadarajan;99] RCH/RCHr](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-13-320.jpg)




![Steps Prediction of Secondary structure (using PSIPRED) Solvent Accessibility Recursive Convex Hull Coordination Number Integration of all these predictions plus other sources of information Final CM prediction (using BioHEL) Using BioHEL [Bacardit et al., 09]](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-23-320.jpg)
![The BioHEL GBML System BIOinformatics-oriented Hiearchical Evolutionary Learning – BioHEL (Bacardit et al., 2007) BioHEL is a rule-based evolutionary learning system that employs the Iterative Rule Learning (IRL) paradigm First used in EC in Venturini’s SIA system (Venturini, 1993) Widely used for both Fuzzy and non-fuzzy evolutionary learning BioHEL inherits most of its components from GAssist [Bacardit, 04], a Pittsburgh evolutionary learning system](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-24-320.jpg)






![1. Three windows of residues Two windows of ±4 residues around the two target amino-acids One window of ±2 residues around the middle point in the chain between the two targets [Punta and Rost, 05] Each position in all three windows contains: PSSM profile (from PSI-BLAST) Predicted SS, SA, RCH and CN](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-31-320.jpg)
![Description of connecting segment and the whole sequence 2. The segments are described by the distribution of Amino acid types Predicted SS, RCH, SA and CN [Punta and Rost, 05] 3. Other information Sequence length Separation between targets Contact propensity between the amino acid types of the targets [Shackelford and Karplus, 07]](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-32-320.jpg)







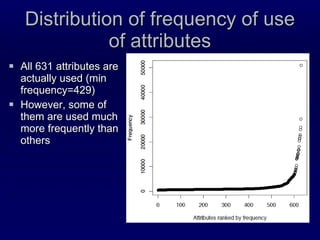
![Rule generated by BioHEL Att PredSS_r1_1 is E,X and Att PredRCH_r1 is 4 Att PredCN_r1_-1 is 0,2,3,4,X and Att PredCN_r2_1 is 3,4 and Att AA_freq_central_P=0 and Att AA_freq_global_E is [0.02,0.10] and Att PSSM_r2_-1_Y is [-7,9.69] and Att PSSM_r2_0_I is [1.76,8] then contact 8 attributes in this rule out of 631 (in average 8.3 att/rule)](https://image.slidesharecdn.com/talk-weizmann-101202094834-phpapp01/85/Data-Mining-Protein-Structures-Topological-Properties-to-Enhance-Contact-Map-Predictions-43-320.jpg)




















































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)


















