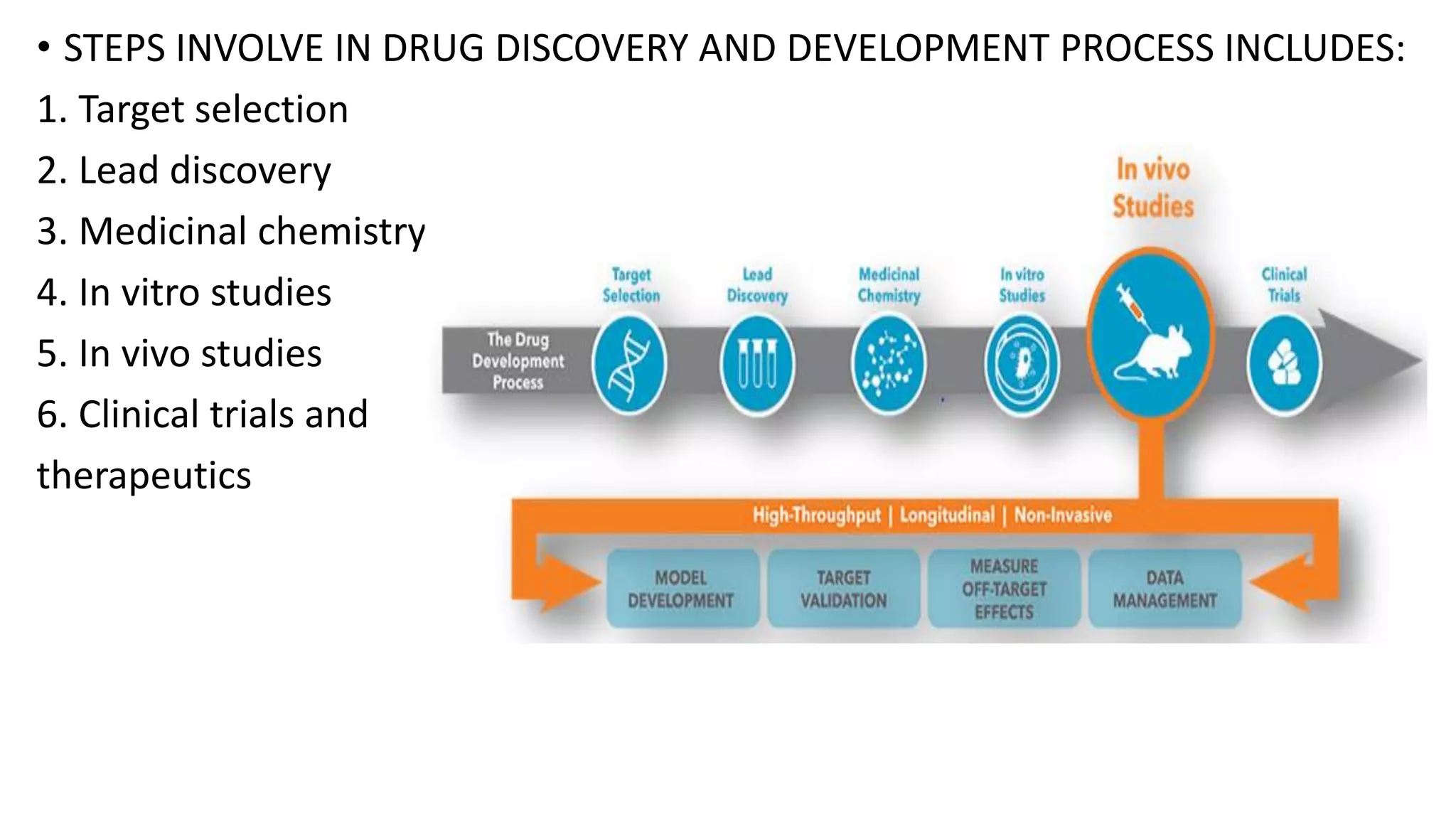
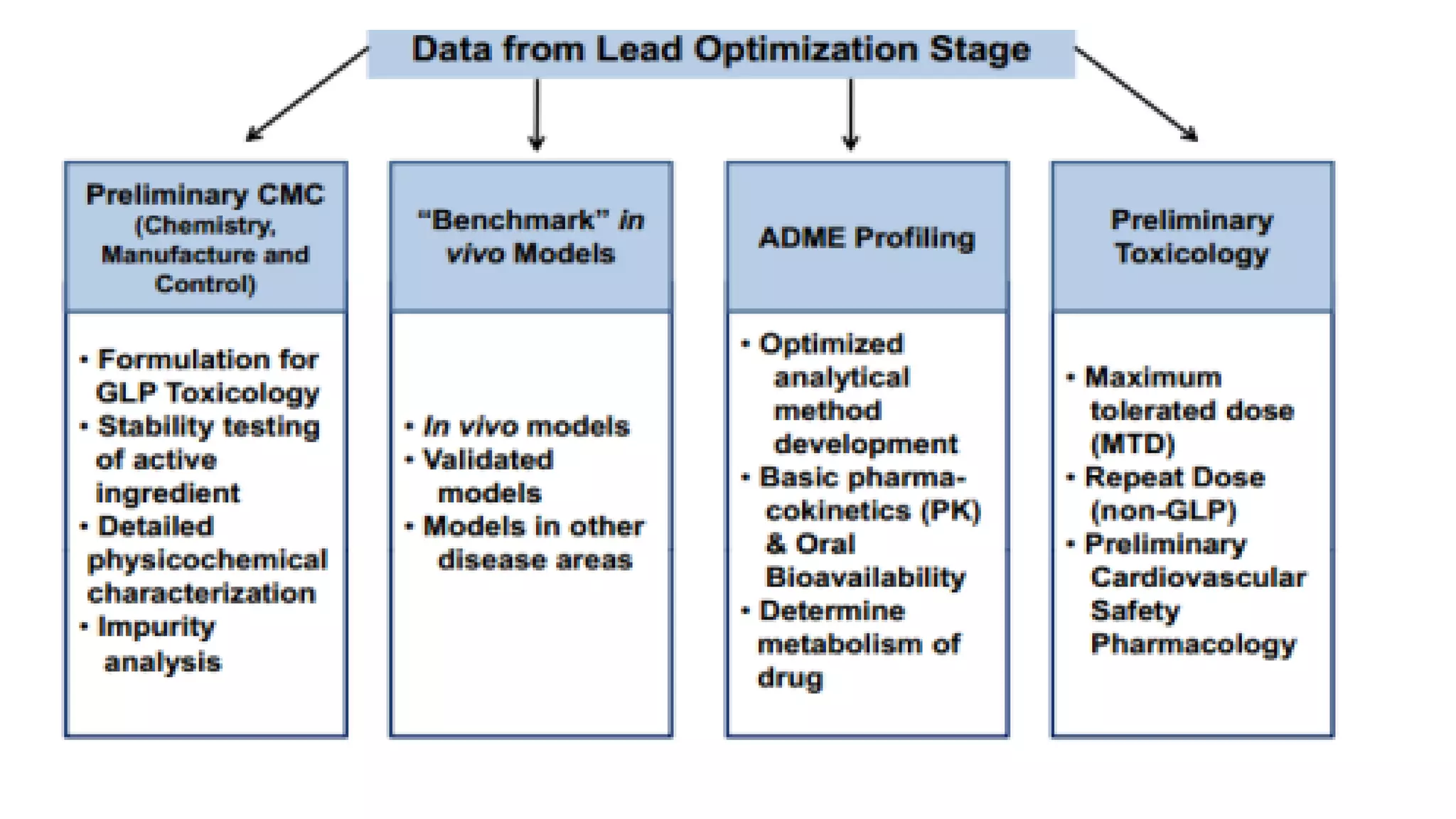
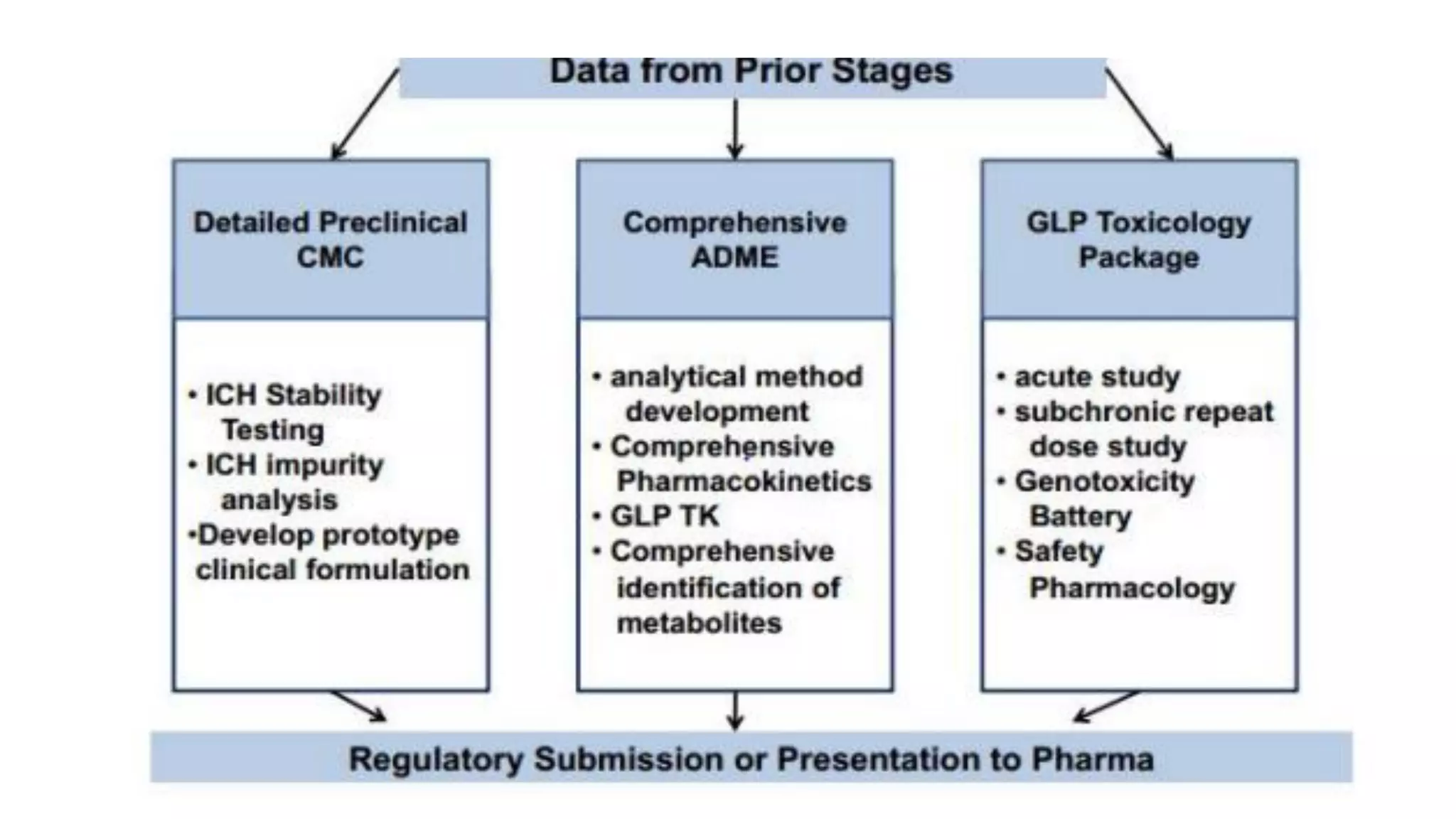
The document discusses computer-based drug design and the drug development process. It describes how computational tools can be used in a multidisciplinary approach to increase efficiency, reduce costs and time, and help design drugs to overcome toxic side effects. The key steps in the drug development process are discussed, including target selection, lead discovery through screening compounds and identifying lead molecules, lead optimization to improve properties, and clinical trials. Computational methods like ligand-based and structure-based drug design are used to model potential drug candidates in the goal of developing safe and effective therapeutics.