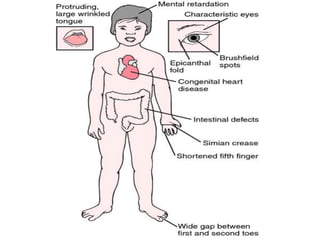
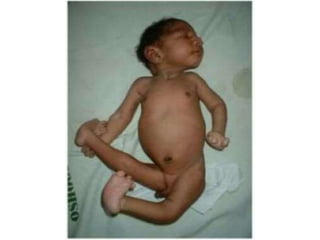
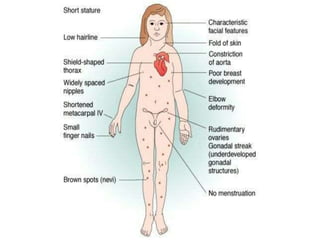
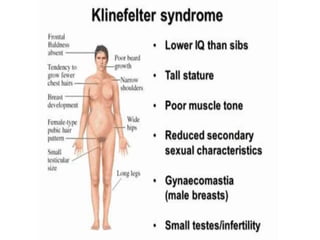
Chromosomal disorders occur in 0.4% of live births and are a significant cause of mental retardation and congenital anomalies, including conditions like Down syndrome (trisomy 21), Edwards syndrome (trisomy 18), and Patau syndrome (trisomy 13). Abnormalities can result from aneuploidy, such as trisomies, or structural defects, and various genetic syndromes can lead to distinct physical traits and health complications. Treatment may involve supportive care and specific interventions for associated conditions; genetic counseling is often recommended due to risks associated with maternal age.

![Chromosomal Abnormalities
• 1-Abnormalities of Chromosomal
Number,
When a human cell has 23 chromosomes, such as human ova or sperm,
it is in the haploid state (n). After conception, in cells other than the
reproductive cells, 46 chromosomes are present in the diploid state
(2n).. Trisomy, an example of aneuploidy, is the presence of three of a
particular chromosome rather than two. It results from unequal division,
called nondisjunction, of chromosomes into daughter cells. Trisomies
are the most common numerical chromosomal anomalies found in
humans (eg, trisomy 21 [Down syndrome], trisomy 18, and trisomy 13)..](https://image.slidesharecdn.com/chromosomaldisorders-220624182230-9235209a/85/CHROMOSOMAL-DISORDERS-2-320.jpg)