This document presents the crystal structure of mature apo-caspase-6, an enzyme involved in neurodegenerative diseases. The structure reveals the canonical caspase conformation, contrasting with a previous structure of apo-caspase-6 that showed a noncanonical conformation. The authors believe the previous structure represented an inactive pH-dependent form. Comparison to other caspase structures allows visualization of loop rearrangements upon ligand binding. This new structure provides insight into the conformational dynamics of caspase-6.



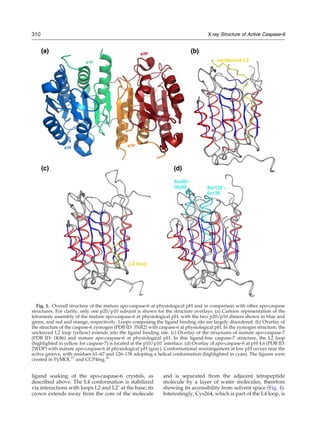
![the tetramer consisting of chains A/B and C/D (Fig.
1a). The overall topology of caspase-6 at pH 7.4
closely resembles the caspase-6 zymogen structure
[Protein Data Bank (PDB) ID: 3NR2; RMSD of 0.74 Å
for 205 Cα
atoms within the p20/p10 dimer; Fig. 1b]
and the mature ligand-free caspase-7 (PDB ID: 1K86;
196 topologically equivalent Cα
atoms within the
p20/p10 dimer superimposing with an RMSD of
0.85 Å; Fig. 1c), with a central six-stranded mixed
β-sheet flanked by five α-helices. It is distinct from
the structure reported for caspase-6 at pH 4.6 (PDB
ID: 2WDP; RMSD of 3.3 Å for 203 Cα
atoms within
the p20/p10 dimer; Fig. 1d), leading to our
hypothesis that crystallization of caspase-6 at low
pH likely sequestered a pH-inactivated form, which
had not been observed when caspase-3 or caspase-7
was crystallized at pH b5 (i.e., PDB IDs: 2C1E and
1KMC29
). We will subsequently refer to the struc-
tures obtained at pH 4.5 and pH 7.4 as low-pH and
physiological-pH mature apo-caspase-6 structures,
respectively.
The caspase-6 protein used for crystallization at
physiological pH comprised residues 24–179 and
194–293 after self-cleavage of the expressed caspase-6
zymogen. Caspase activity was confirmed prior to
crystallization (Fig. 2). The electron density is well
defined for residues Phe31-Cys163 in the p20
subunit, residues Tyr198-Val212 and Thr222-
Arg260, and residues Gln274-Lys291 in the p10
subunit. Consequently, the first seven residues of
the N-terminus of the p20 subunit, as well as the last
two residues of the p10 subunit, were absent from
the electron density maps, and the loops constitut-
ing the catalytic grooves L2 (residues 163–179), L3
(212–222), and L4 (257–275) were found to be
disordered.
The L2′ loop
For inhibitor-bound caspase-3 and caspase-7, the
conformation of the loops forming the catalytic
groove has been shown to be stabilized by
interactions with the cleaved interdomain linker
loop L2′ of the adjacent p20/p10 dimer;30,31
however, in the structure of the pro-caspase-7
zymogen and the ligand-free caspase-7, the L2′
loop is folded back and located at the interface
between the two p10 subunits of the p202/p102
tetramer.32,33
In the apo-caspase-6 structure at
physiological pH, the L2′ loop is also oriented
towards the p10/p10′ interface (Fig. 3). It has
been proposed that the presence of inhibitor or
substrate triggers flipping of the L2′ loop.34
In
support of this hypothesis, apo-crystal structures
of caspase-3 and caspase-7 obtained in the
presence of inhibitor35,36
showed that the L2′
loop folded against the L2 and L4 loops. This was
in contrast to the zymogen-like conformation of
the L2′ loop in the previously reported apo-
caspase-7 structure crystallized in the absence of
inhibitor.33
These studies on caspase-3 and cas-
pase-7 suggest ligand-induced dynamics of the
L2′ loop. Our crystallography-based insight into
the mature apo-caspase-6 and literature evidence
further support the presence of multiple apostruc-
tures that are likely to result from the inherent
conformational freedom of the enzyme and could
be stabilized by ligand binding. In the apo-
caspase-6 crystals, the ligand binding site is
located at a large solvent channel through the
crystal and should be accessible to the ligand via
soaking. Furthermore, the conformation of the L4
loop is not restricted by crystal packing; thus,
rearrangement upon ligand binding should be
feasible. Based on this insight, we soaked an
inhibitor into the apo-caspase-6 crystals. Unfortu-
nately, this approach failed; specifically, longer
soaking times resulted in complete crystal decay,
whereas shorter exposure to the molecule afforded
low compound occupancy. We propose that
within the crystal packing context, the L2′ loop
in its locked position at the dimer interface is
not able to reorient towards, and to stabilize,
the L4 loop in a conformation required for
ligand binding, as observed in the Ac-VEID-CHO
caspase-6 complex. (Further details can be found
in Supplementary Material.)
Comparison with the Ac-VEID-CHO caspase-6
complex
In the Ac-VEID-CHO caspase-6 complex (PDB ID:
3OD523
), the L2′ loop is flipped by 180° and oriented
towards the L2/L4 loop interface of the neighboring
p20/p10 dimer (Fig. 3). The conformation of loops L2
and L3 closely resembles the one observed after the
Table 1. Data processing and refinement statistics
Parameter Apo-caspase-6
PDB ID 3P45
Space group P21
Cell dimensions
a, b, c (Å) 81.23, 161.24, 88.92
α, β, γ (°) 90.0, 94.80, 90.0
Resolution (Å) 30–2.53
Rmerge
a
(%) 10.1 (55.0)
Mean I/σIa
8.0 (2.0)
Completenessa
(%) 99.8 (99.8)
Multiplicity 2.9
Refinement
Number of reflections 75,784
Rwork/Rfree (%) 20.7/26.4
B-factors
Protein 10.3
Water 8.7
RMSD
Bond lengths (Å) 0.019
Bond angles (°) 1.742
a
Values in parentheses are for the highest-resolution shell.
309X-ray Structure of Active Caspase-6](https://image.slidesharecdn.com/e0c9e0d4-a6d3-4d52-aebb-e3a04c198c7d-150530213614-lva1-app6892/85/casp6-paper-JMB2011-3-320.jpg)

![In conclusion, this article describes the structures
of caspase-6 in its apo form at physiological pH. It
closely resembles mature ligand-free caspase-7 with
a central six-stranded mixed β-sheet flanked by five
α-helices. Loops L2, L3, and L4, which constitute the
ligand binding site in the Ac-VEID-CHO caspase-6
complex, are disordered, and the L2′ loop resides at
the p10/p10′ interface as observed in the caspase-6
zymogen structure. Our results indicate that the
presence of ligand induces loop L2′ to reorient
towards, and to stabilize, the L2 and L4 loops of the
neighboring p20/p10 dimer, with the L2′ flip
required for ligand binding. The overall topology
of the apostructure at physiological pH is distinct
from the structural data previously reported for
caspase-6 at pH 4.5. Even though its noncanonical
fold does not impair inhibitor binding, we think that
it does not represent a physiologically relevant
conformation that would provide an allosteric site
for drug development.
Materials and Methods
Gene construction and protein expression
Oligonucleotides were designed to amplify the cata-
lytic domain (amino acid residues 24–293) of human
caspase-6 for cloning into a BioFocus in-house expres-
sion vector. cDNA products of the correct size for
caspase-6 were obtained from polymerase chain reaction
experiments using cDNA encoding full-length caspase-6.
Products were successfully cloned into the BioFocus
vector pT7-CH for Escherichia coli expression under the
control of the T7 promoter. The vector–insert combina-
tion provided a C-terminal hexa-histidine purification
tag on the expressed protein. For large-scale expression,
recombinant human caspase-6 was expressed in Rosetta
E. coli cells grown overnight at 37 °C from glycerol
stock. The next day, 6×1 L cultures were inoculated
with 50 ml of overnight starter cultures. Cells were
grown at 37 °C until an OD600 of 1.2 had been reached.
Cells were cooled to 25 °C, induced with 0.5 mM IPTG,
and shaken overnight before being harvested. Cell
pellets were harvested, washed with phosphate-buffered
saline, and then stored at −80 °C. We found that the
temperature had clearly an effect on the yield of fully
processed caspase-6 protein, as induction at 25 °C,
compared to 37 °C, resulted in higher yields. In addition,
increased protein yields were also obtained when the
cells were induced at a higher cell density.
Protein purification
Cell paste from 6×1 L cultures was resuspended on ice
in 100 ml of 25 mM Tris (pH 8.0) containing 25 mM
imidazole, 100 mM NaCl, 10% glycerol, and 0.1% 3-[(3-
cholamidopropyl)dimethylammonio]propanesulfonic
acid. Following mechanical disruption of the cells, the
soluble fraction was harvested by centrifugation at
60,000g for 30 min. The cleared supernatant was incubated
batchwise with 0.5 ml of NiNTA (Qiagen) for 2 h at 4 °C to
allow binding. The resin was collected by centrifugation at
400g and washed once with buffer A [25 mM Tris (pH 8.0)
containing 25 mM imidazole, 100 mM NaCl, 10% glycerol,
and 0.1% 3-[(3-cholamidopropyl)dimethylammonio]pro-
panesulfonic acid] before being loaded into an Omnifit
column. The column was attached to an ÄKTAexpress,
run through IMAC elution (using buffer A supplemented
with 500 mM imidazole and 10 mM DTT), and then
subjected to size-exclusion chromatography [with a
Superdex 200 16/60 column equilibrated in 20 mM
sodium acetate (pH 5.5) containing 50 mM NaCl and
10 mM DTT], as the protein was most stable for storage
under these conditions. After size-exclusion chromatog-
raphy, the fractions were collected and analyzed by SDS-
PAGE stained with Coomassie brilliant blue. The purest
fractions were pooled, concentrated to 8.2 mg/ml by
ultrafiltration, and used for crystallization. DTT (10 mM)
was added to the concentrated protein prior to crystalli-
zation. Protein concentration was determined with
Fig. 4. Comparison of inhibitor-bound caspase-3, caspase-6, and caspase-7 structures. Interaction of the L4 loop
residues with the P4 site of bound peptide in the Ac-VEID-CHO caspase-6 (PDB ID: 3OD5; gray), Ac-DEVD-CHO
caspase-3 (PDB ID: 2H5I; magenta), and Ac-DEVD-CHO caspase-7 (PDB ID: 1F1J; blue) complexes, respectively. In
contrast to the caspase-3 and caspase-7 complexes, all hydrogen bonds between the L4 loop and the inhibitor peptide are
water mediated in the caspase-6 complex. The figures were created in PyMOL.27
312 X-ray Structure of Active Caspase-6](https://image.slidesharecdn.com/e0c9e0d4-a6d3-4d52-aebb-e3a04c198c7d-150530213614-lva1-app6892/85/casp6-paper-JMB2011-6-320.jpg)
![Coomassie Plus reagent, measuring optical absorbance at
595 nm.
Protein activity
Caspase-6 activity was confirmed using an assay that is
based on the protolytic cleavage of a fluorogenic substrate.
This substrate consists of two caspase-6 recognition
peptide molecules that are covalently linked to two
amine groups of the fluorescent dye rhodamine 110
(Z-VEID-R110; Invitrogen), which suppresses R110 fluo-
rescence. During proteolysis, both recognition peptides
are cleaved off, and subsequent dequenching of the dye
indicates enzymatic activity. Pipes (20 mM; pH 7.4),
100 mM NaCl, 0.03% Pluronic®, 10% sucrose, 1 mM
ethylenediaminetetraacetic acid, and 5 mM glutathione
were used as assay buffer. The enzyme concentration was
determined with Coomassie Plus reagent, measuring
optical absorbance at 595 nm. Recombinant caspase-6
activity was detected by measuring the R110 release from
Z-VEID-R110 at 37 °C using a Perkin-Elmer EnVision® at
an excitation wavelength of 485±14 nm and at an
emission wavelength of 535±25 nm. The enzyme prepa-
ration used in the enzymatic studies was titrated using the
substrate Z-VEID-R110 and was found to be active (results
not shown). The inhibitor concentration that results in 50%
inhibition (IC50) was determined for the reported caspase-
6-competitive and caspase-6-reversible inhibitor Ac-VEID-
CHO.39,40
The inhibitor was resuspended in dimethyl
sulfoxide, serially diluted in assay buffer, and combined
with 1 pg of caspase-6 on a 96-well plate. The maximum
dimethyl sulfoxide concentration was 1%, and the
caspase-6 substrate was used at a concentration of 10 μM.
Crystallization and data collection
Crystals of mature apo-caspase-6 were obtained with
hanging-drop vapor diffusion on 24-well plates (VDXm;
Hampton Research) at 20 °C by mixing 1.0 μl of protein
solution [in 20 mM sodium acetate (pH 5.5), 50 mM NaCl,
and 0.5 mM Tris(hydroxypropyl)phosphine] with 1.0 μl of
reservoir solution (0.5 ml) consisting of 3.3 M sodium
nitrate, 0.1 M Tris (pH 7.4), 0.5% ethyl acetate, and 5 mM
Tris(hydroxypropyl)phosphine. Single crystals
(0.5 mm×0.3 mm×0.1 mm) grew by microseeding
straight into the drop after setup within 1 week at 20 °C.
The crystallization drop was overlaid with 3 μl of
cryoprotectant containing 20% ethylene glycol, 3.5 M
sodium nitrate, and 0.1 M Tris–HCl (pH 7.4), and a crystal
was harvested for data collection. The crystal was flash
frozen in liquid nitrogen, and X-ray data collection was
carried out at 100 K on a Rigaku R-Axis IV image plate
detector, with data indexed, integrated, and scaled using
MOSFLM and SCALA (CCP4),41–43
respectively.
Structure solution and refinement
Chain A of the pH-inactivated caspase-6 model com-
prising residues 31–293, representing one p20/p10 dimer,
was used for molecular replacement using Phaser,44
locating eight p20/p10 subunits in the asymmetric unit.
The resulting model was given two rounds of atomic
refinement with tight geometric weights using
REFMAC5.45
The electron density maps calculated after
molecular replacement and initial refinement were exam-
ined, and residues with a poor fit to the electron density
map were omitted from the model. The truncated model
was used as a starting model for automated model
building in Buccaneer.46
Possibly due to the disorder of
a large number of surface loops and hence chain breaks
around the active site, automated model building failed to
improve the initial model. In an alternative approach,
NCS-averaged electron density maps were calculated in
PARROT47 and used to rebuild missing residues manually
in Coot.48
The resulting extended model was then refined
using REFMAC5. To account for differences in thermal
motion, we performed TLS refinement on the completed
model, with one TLS group per p10/p20 heterodimer.
Water molecules were then added using the water
placement option in Coot and refined using REFMAC5.
For all eight heterodimer molecules in the asymmetric
unit, chain breaks are observed between ~Ala162 and
Tyr198, between ~Val212 and Thr222, and between
~Arg260 and Gln274, and the residues were omitted
from the model. Structural geometry was checked using
PROCHECK and MolProbity.49,50
Accession number
Atomic coordinates and structure factors have been
deposited in the PDB under accession code 3P45.
Supplementary Data
Supplementary data associated with this article
can be found, in the online version, at doi:10.1016/
j.jmb.2011.05.020
References
1. Li, J. & Yuan, J. (2008). Caspases in apoptosis and
beyond. Caspases in apoptosis and non-apoptotic
processes. Oncogene, 27, 6194–6206.
2. Lavrik, I. N., Golks, A. & Krammer, P. H. (2005).
Caspases: pharmacological manipulation of cell death.
J. Clin. Invest. 115, 2665–2672.
3. Mykles, D. L. (2001). Proteinase families and their
inhibitors. Methods Cell Biol. 66, 247–287.
4. Earnshaw, W. C., Martins, L. M. & Kaufmann, S. H.
(1999). Mammalian caspases: structure, activation,
substrates, and functions during apoptosis. Annu.
Rev. Biochem. 68, 383–424.
5. Thornberry, N. A. & Lazebnik, Y. (1998). Caspases:
enemies within. Science, 281, 1312–1316.
6. de Calignon, A., Fox, L. M., Pitstick, R., Carlson, G. A.,
Bacskai, B. J., Spires-Jones, T. L. & Hyman, B. T. (2010).
Caspase activation precedes and leads to tangles.
Nature, 464, 1201–1204.
7. Guo, H., Albrecht, S., Bourdeau, M., Petzke, T.,
Bergeron, C. & LeBlanc, A. C. (2004). Active caspase-6
and caspase-6-cleaved tau in neuropil threads, neuritic
plaques, and neurofibrillary tangles of Alzheimer's
disease. Am. J. Pathol. 165, 523–531.
313X-ray Structure of Active Caspase-6](https://image.slidesharecdn.com/e0c9e0d4-a6d3-4d52-aebb-e3a04c198c7d-150530213614-lva1-app6892/85/casp6-paper-JMB2011-7-320.jpg)




















![apoptosis new 1 [Autosaved] cell biology.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/apoptosisnew1autosaved-251012171439-ca8ede0f-thumbnail.jpg?width=640&height=640&fit=bounds)





























