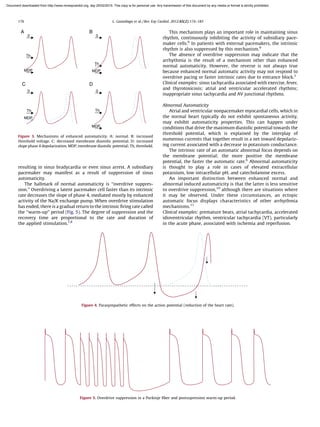
This document summarizes the normal mechanisms of cardiac cellular electrophysiology and the three main mechanisms responsible for cardiac arrhythmias: automaticity, triggered activity, and reentry. It describes how cardiac myocytes generate action potentials through the coordinated flow of ions like sodium, calcium, and potassium. Automaticity refers to the ability of pacemaker cells to spontaneously depolarize. Triggered activity occurs when early or delayed afterdepolarizations trigger premature action potentials. Reentry involves abnormal conduction that allows impulse propagation to reactivate previously excited tissue. Understanding these mechanisms is important for properly diagnosing and treating different types of arrhythmias.