Downloaded 84 times







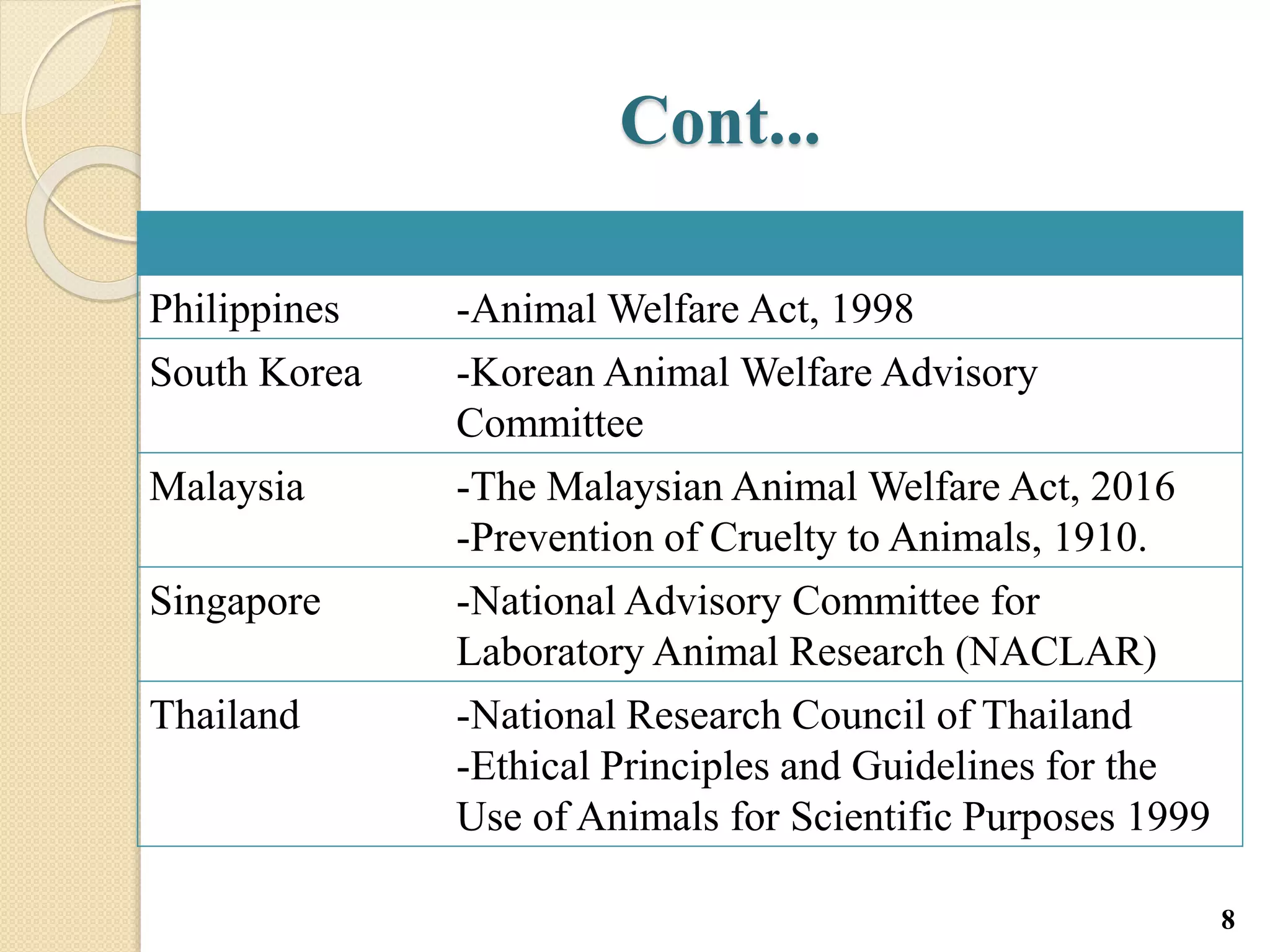






















The document discusses animal testing, outlining its rationale, advantages, and disadvantages, and examines welfare initiatives in several Asian countries. It emphasizes the importance of animal testing in advancing medical research and drug discovery, while addressing ethical considerations and the need for adherence to CPCSEA guidelines to minimize animal suffering. Additionally, it details the care and maintenance of laboratory animals to ensure their well-being during experimentation.











![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)































































