Downloaded 102 times












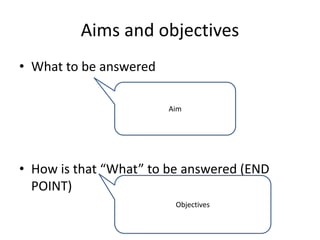




















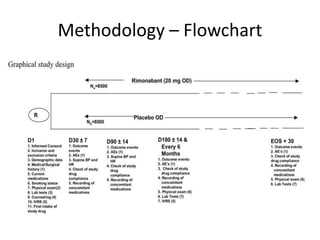
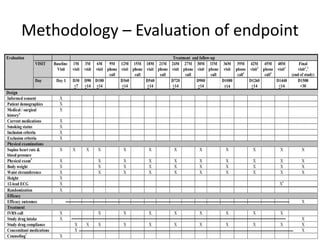
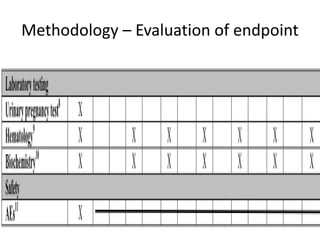


























This document provides guidelines for developing a clinical trial protocol including general information, background, aims and objectives, inclusion/exclusion criteria, study design, methodology, statistics, ethics, quality control, and protocol amendments. Key elements that must be addressed in the protocol are enumerated such as the research question, treatment characteristics, data collection and analysis, and legal responsibilities. Protocol deviations and violations that occur during the study are also discussed along with examples and regulatory requirements for reporting them. Adherence to good clinical practice and obtaining necessary approvals are emphasized.



































































