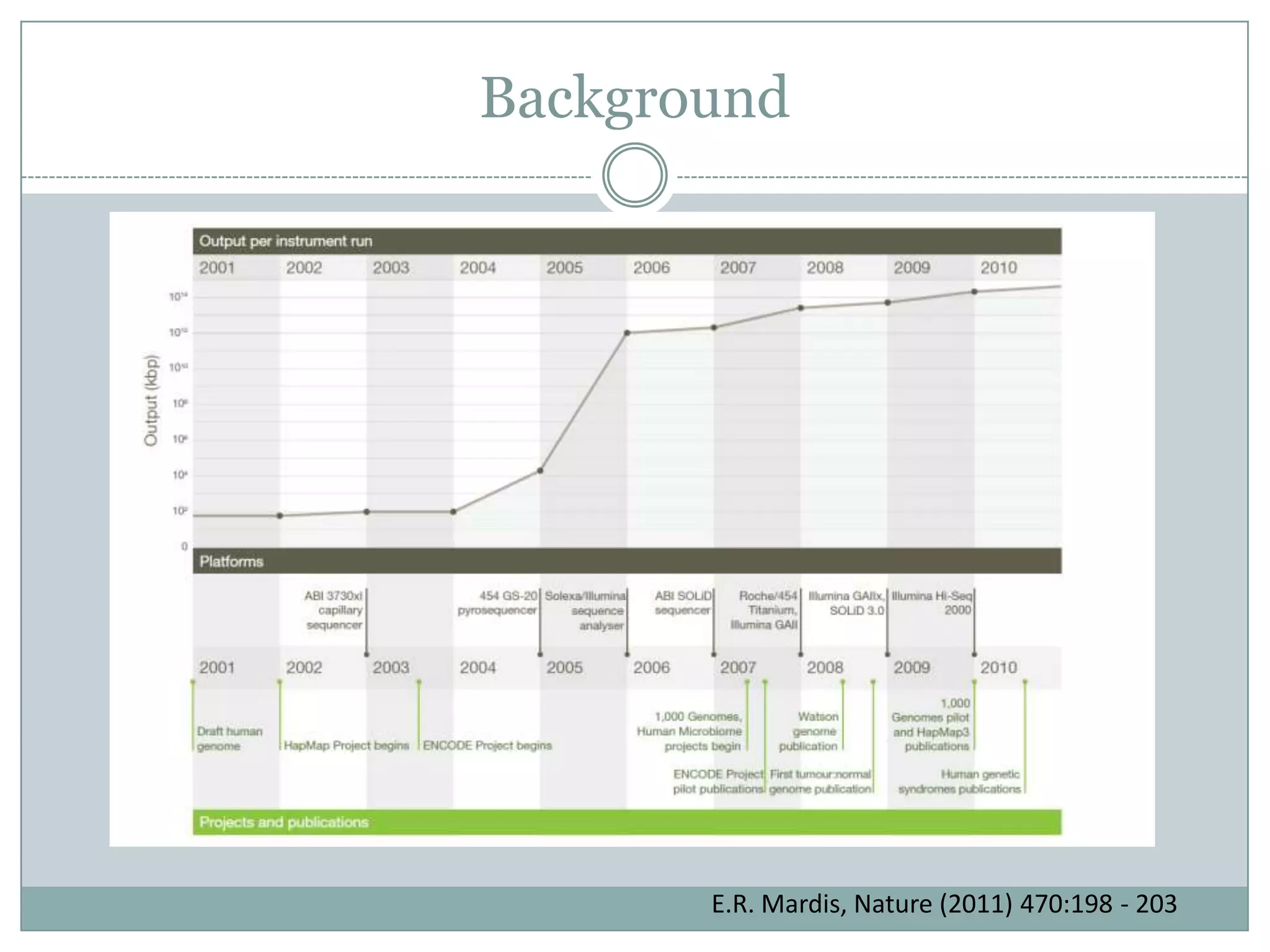
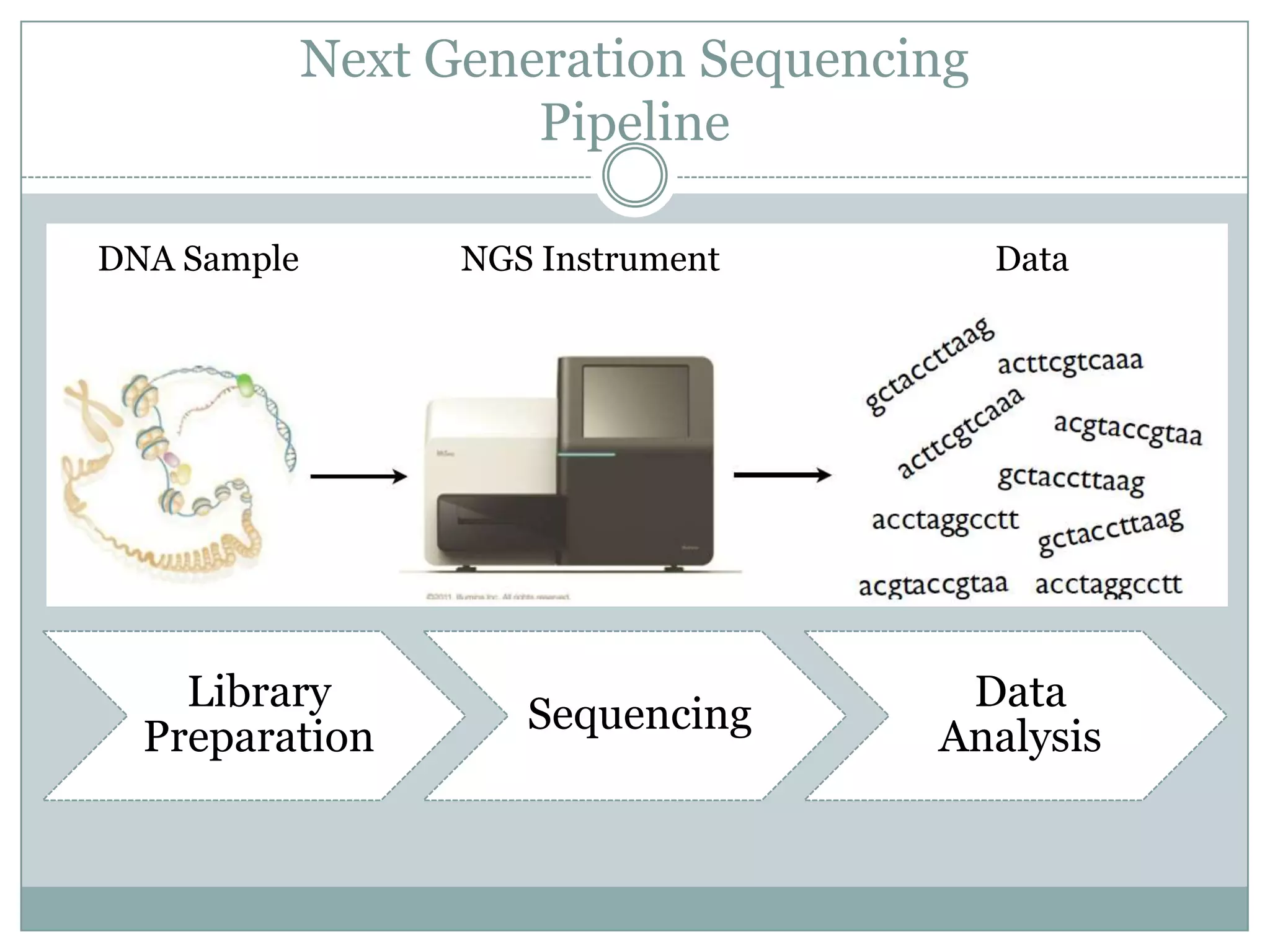
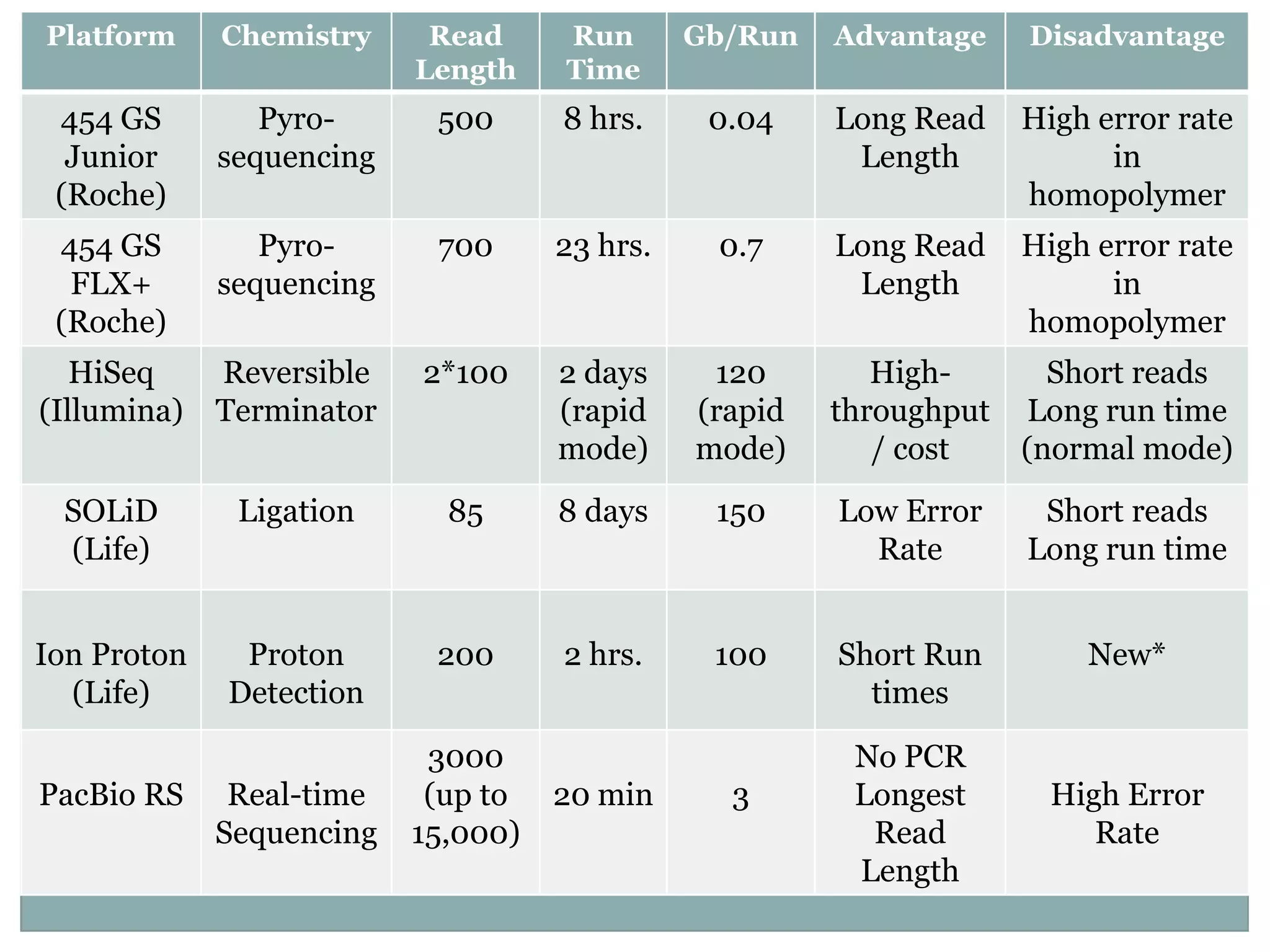
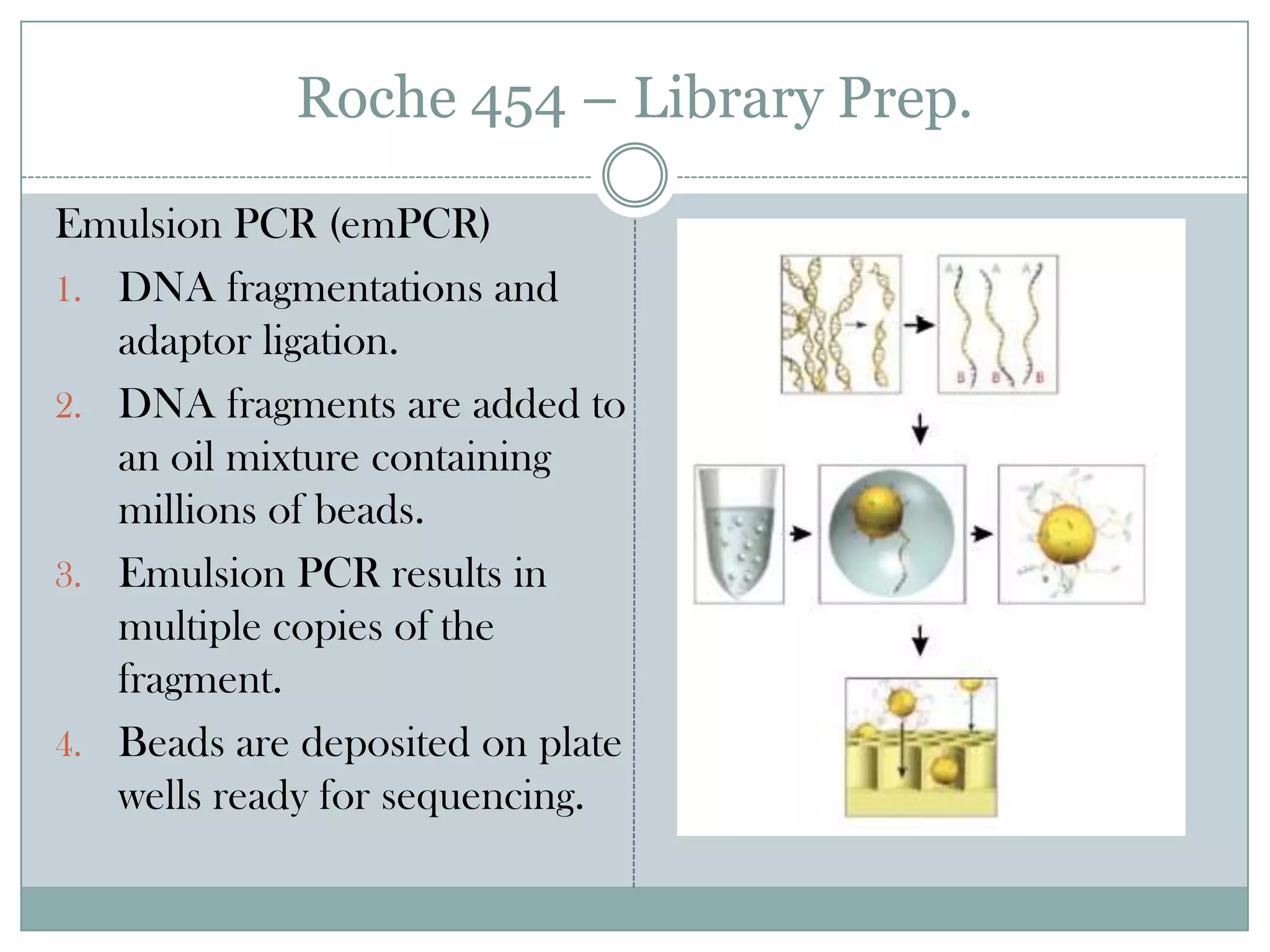
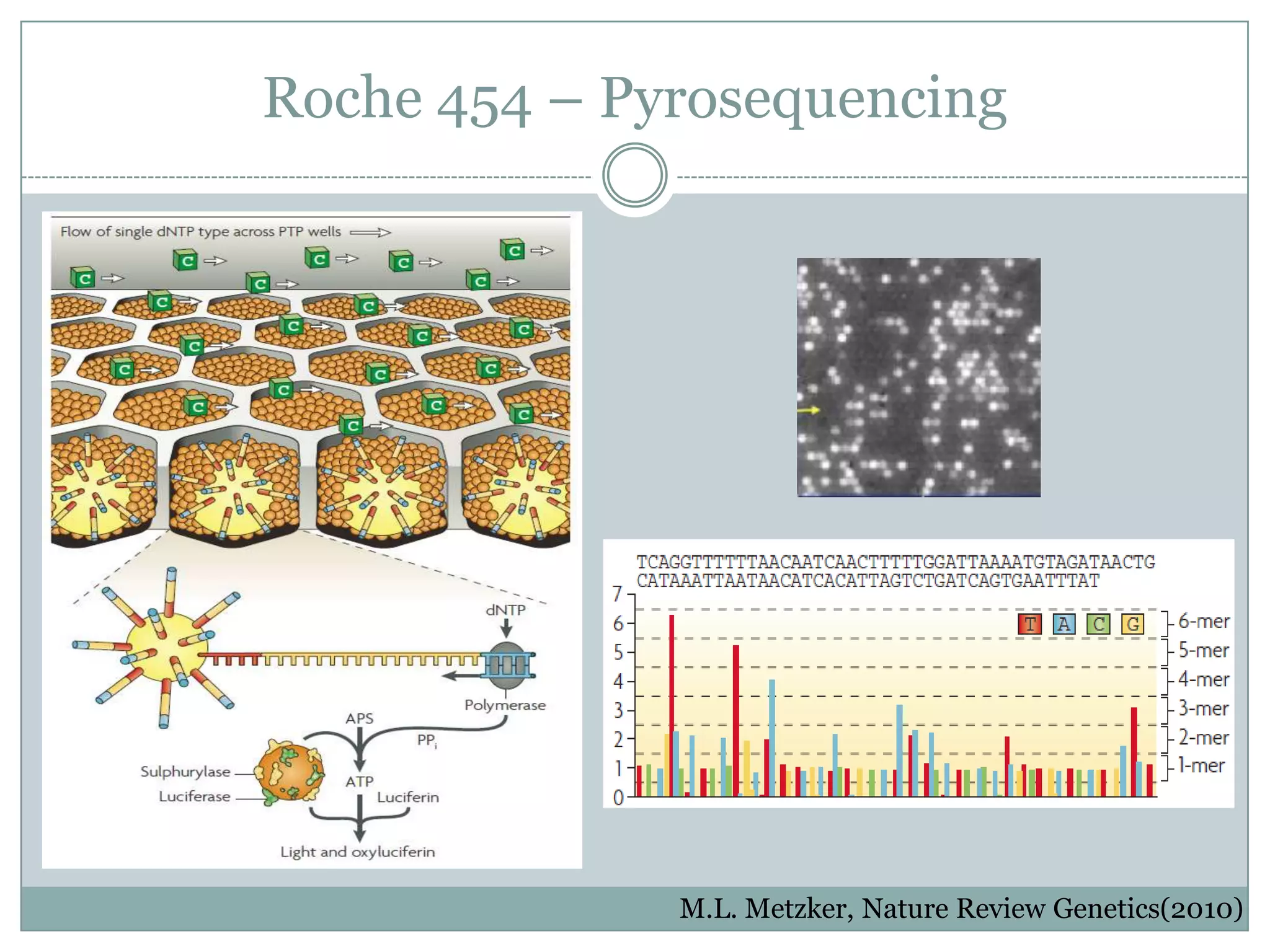
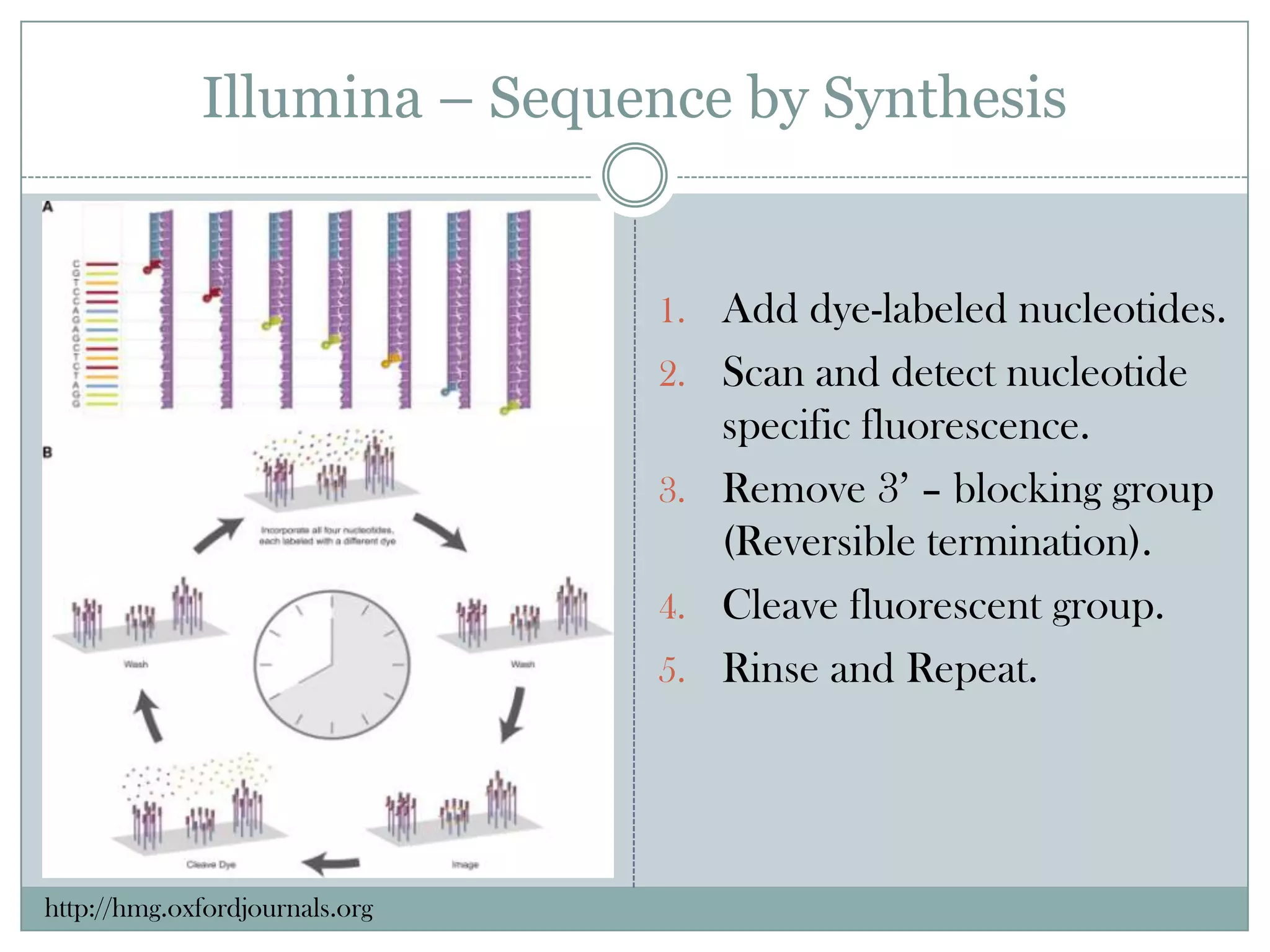
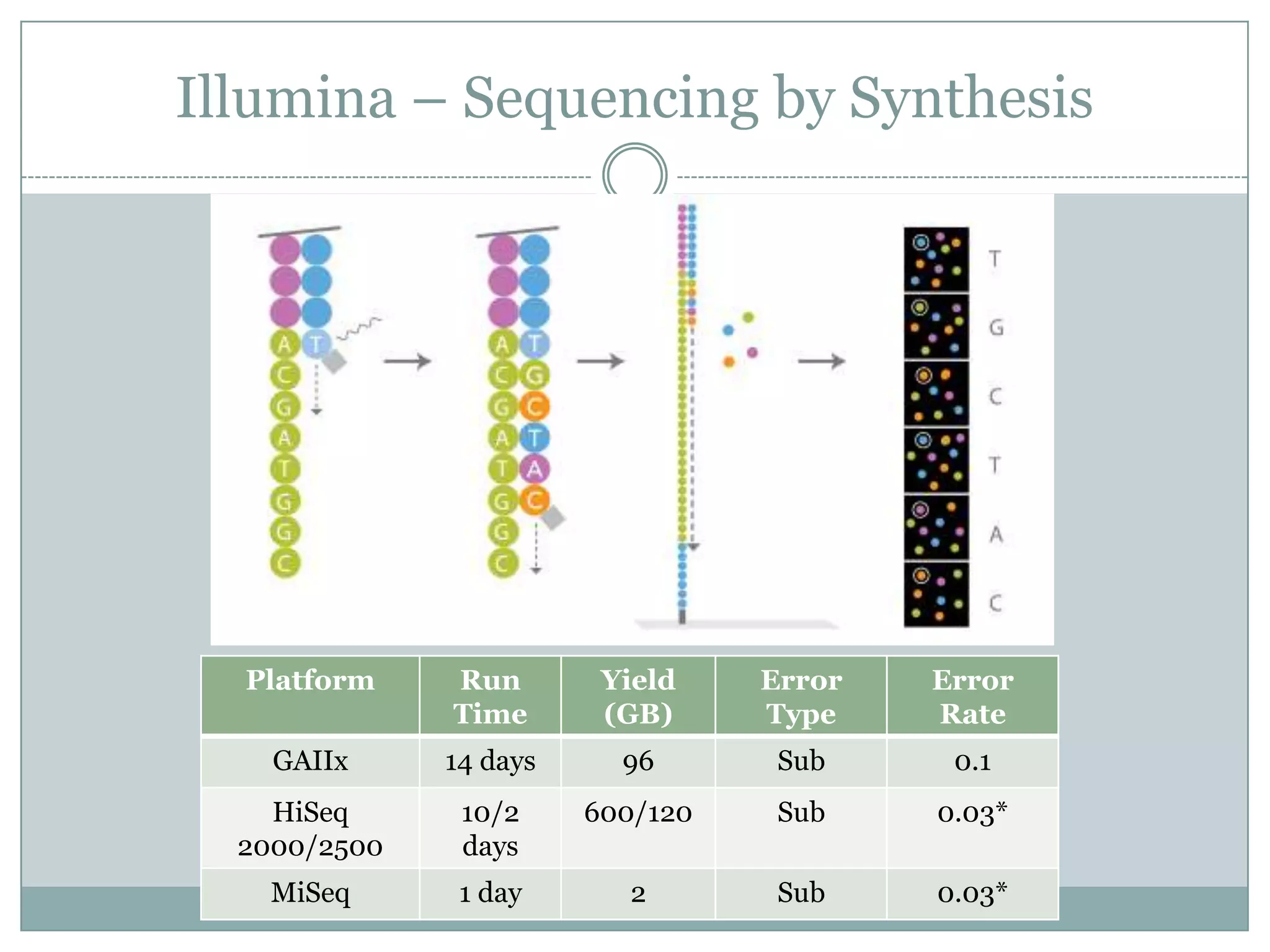
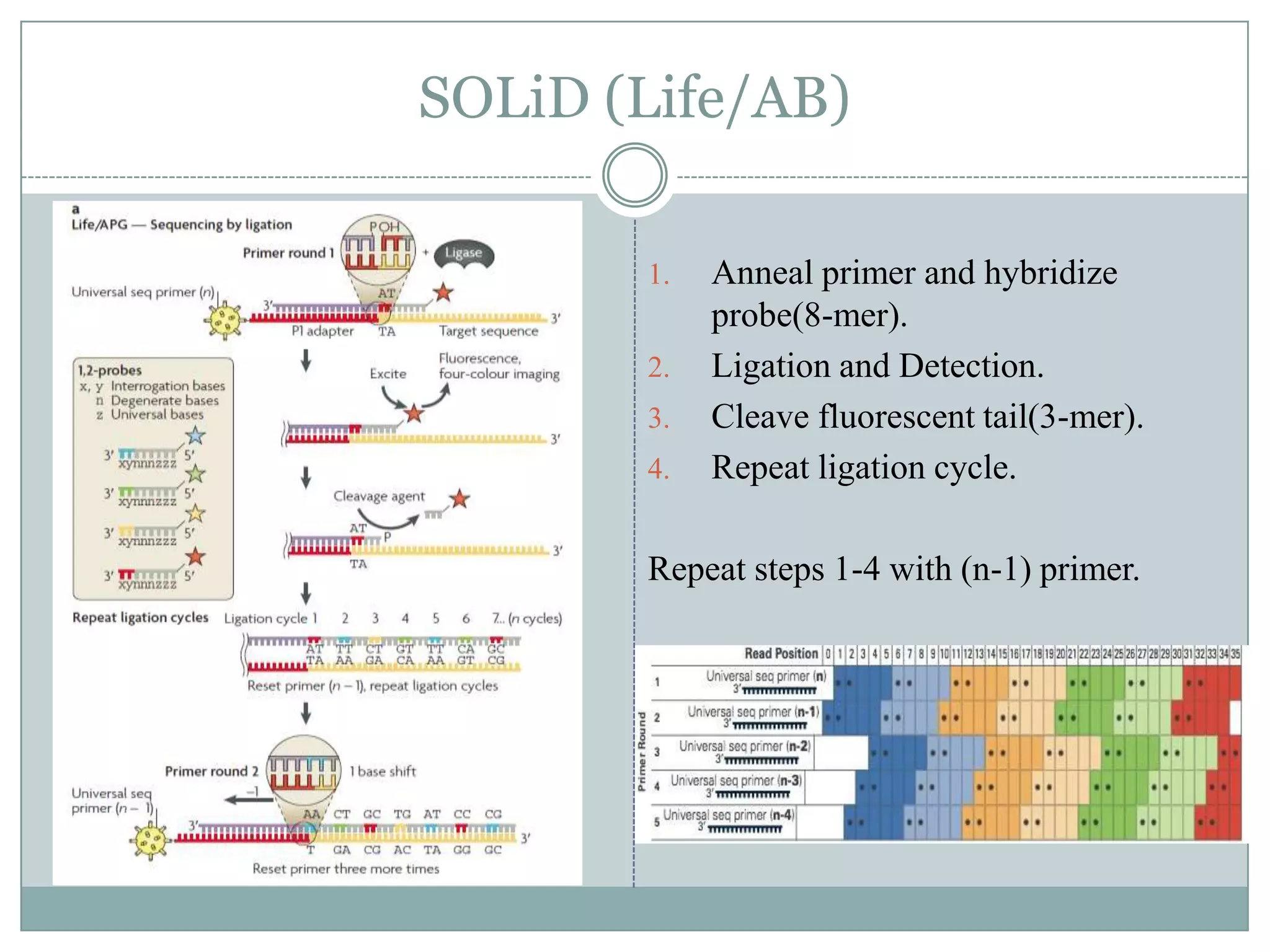
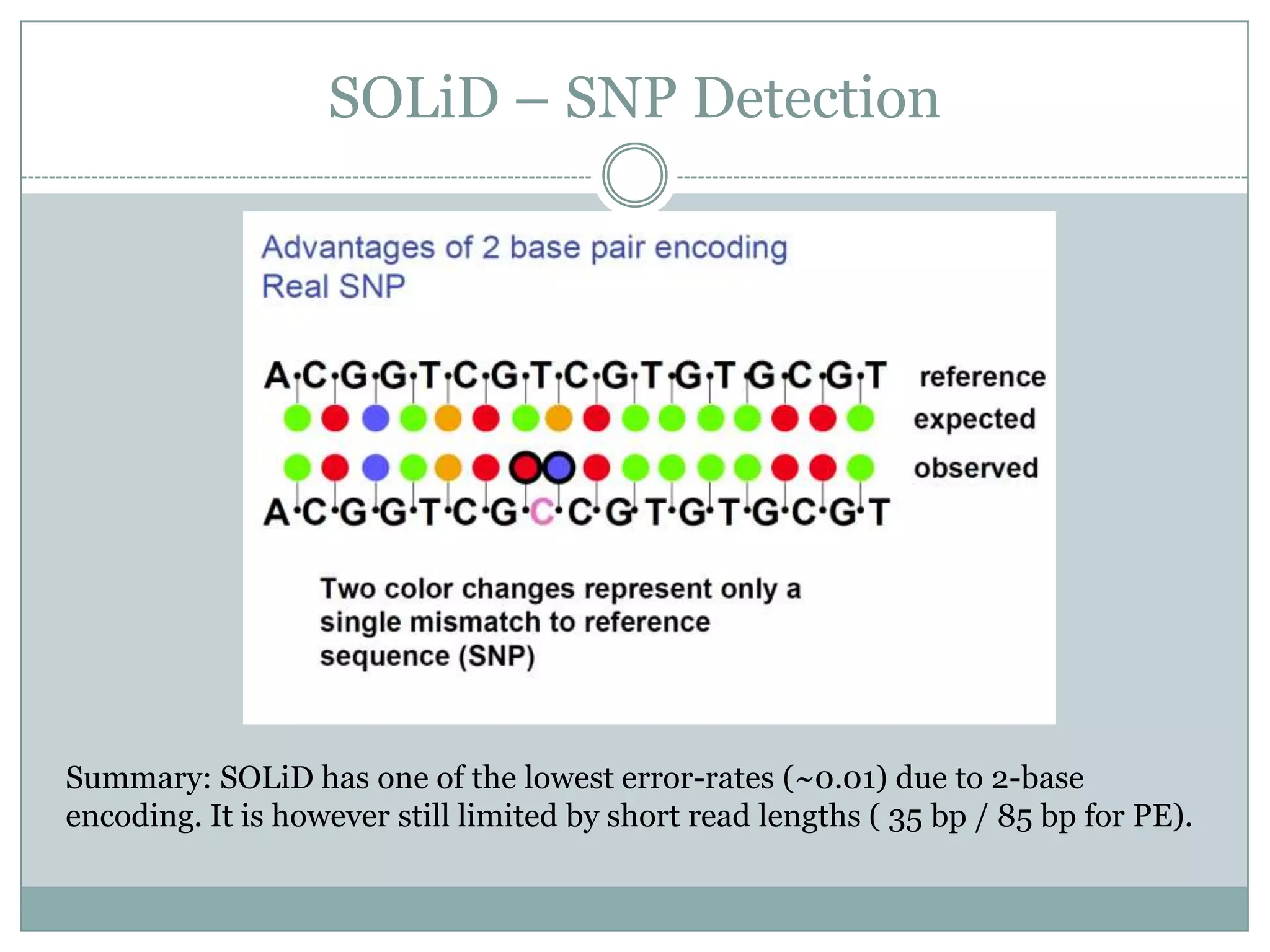
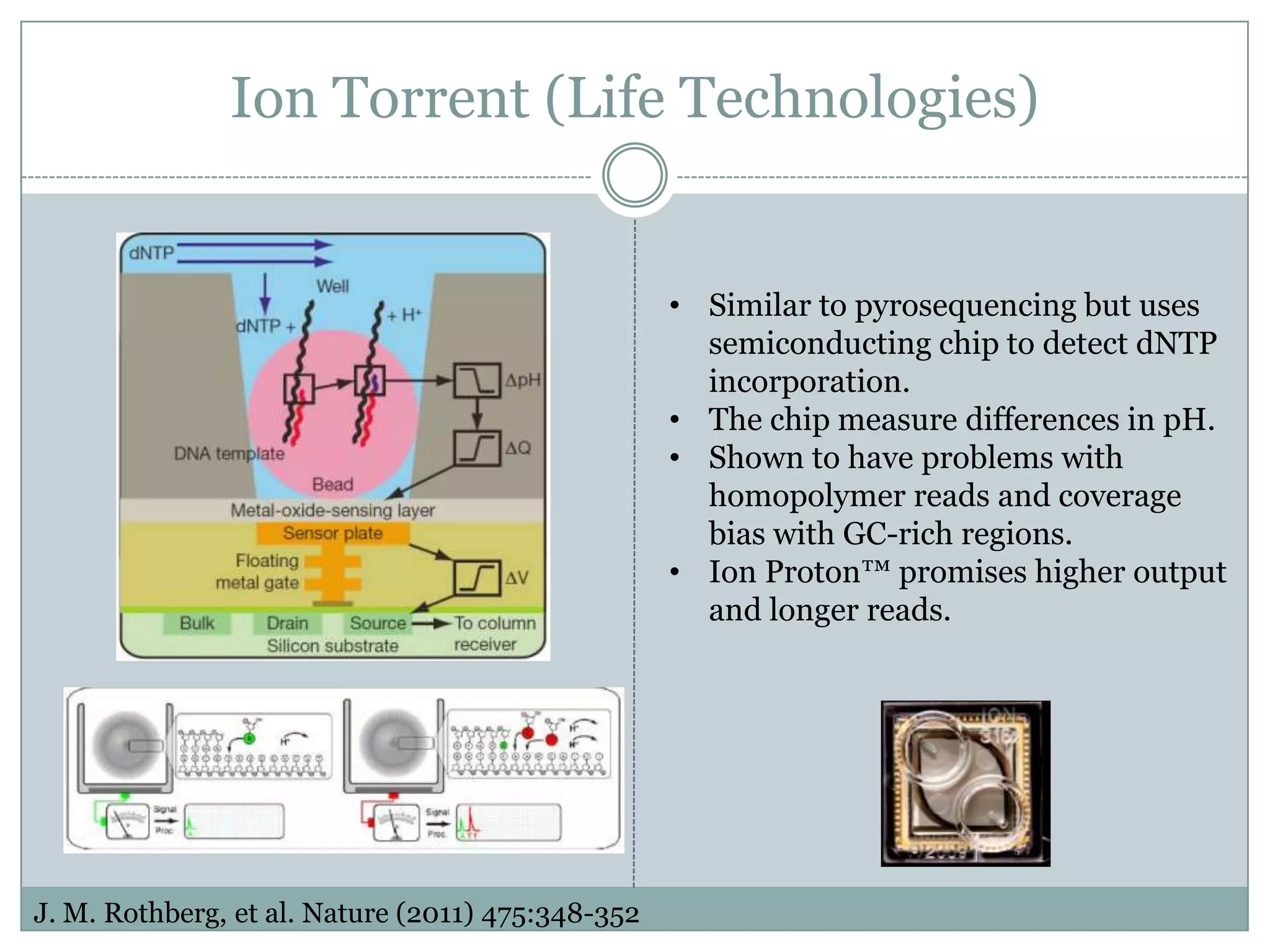
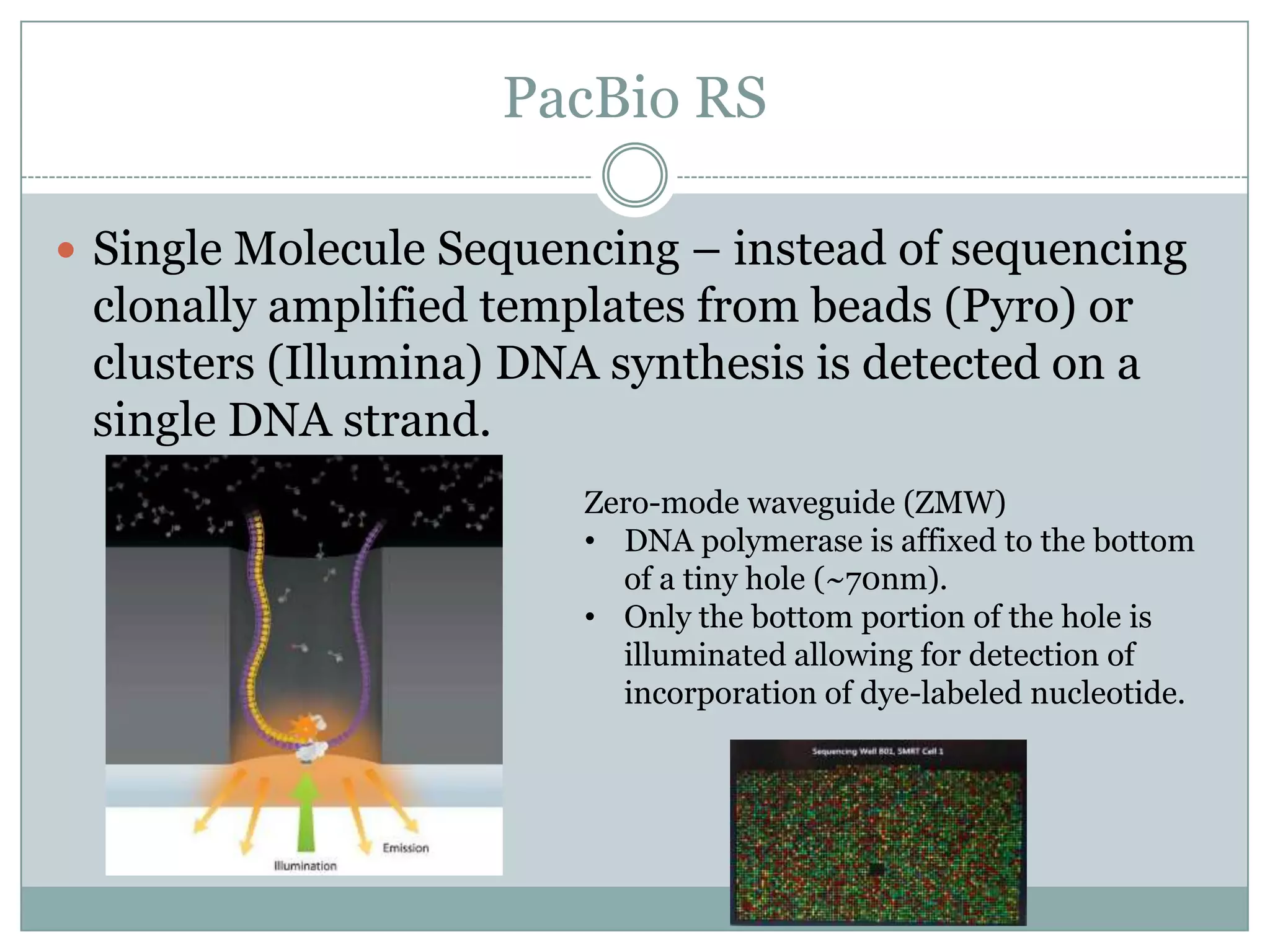
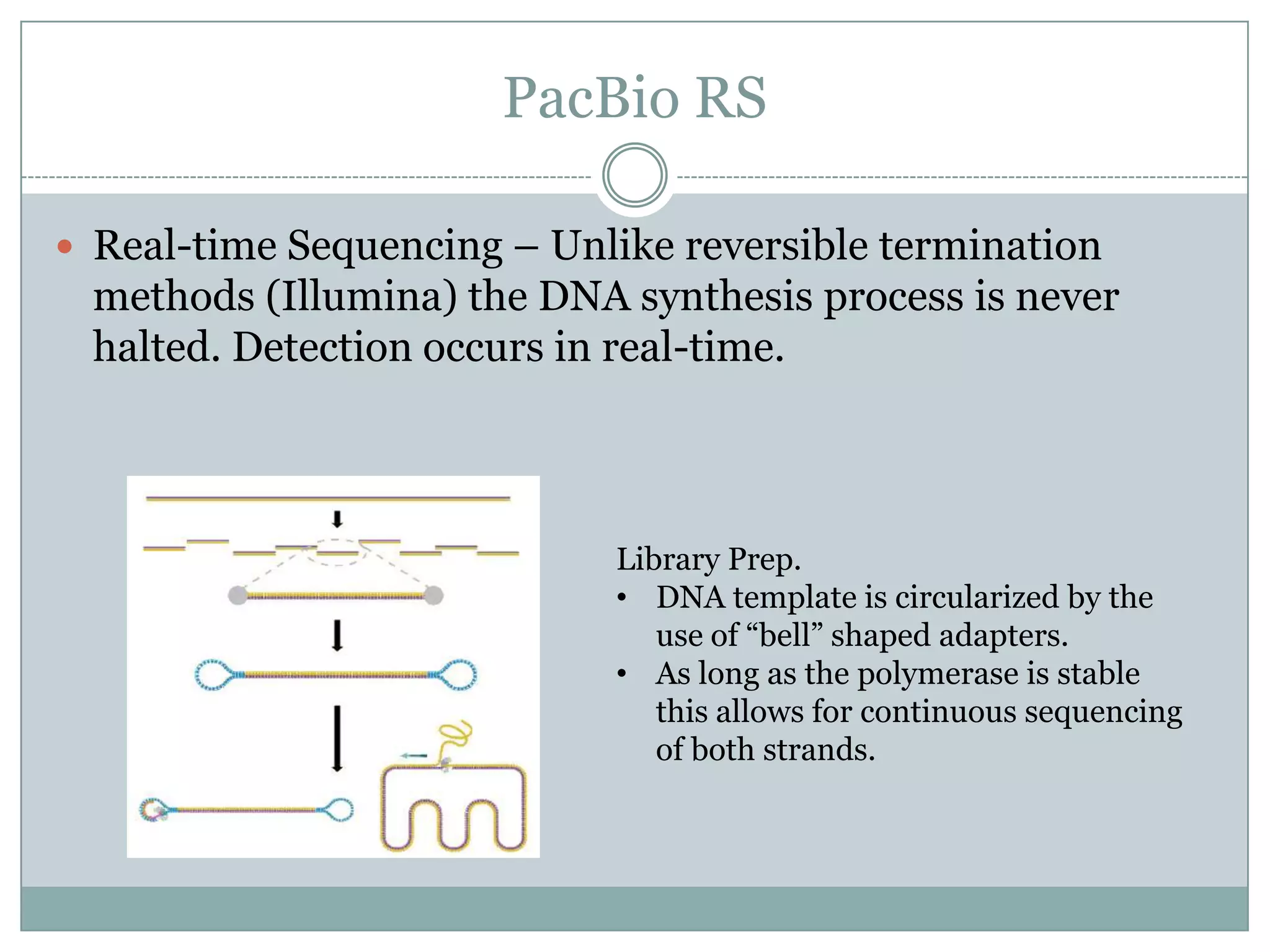
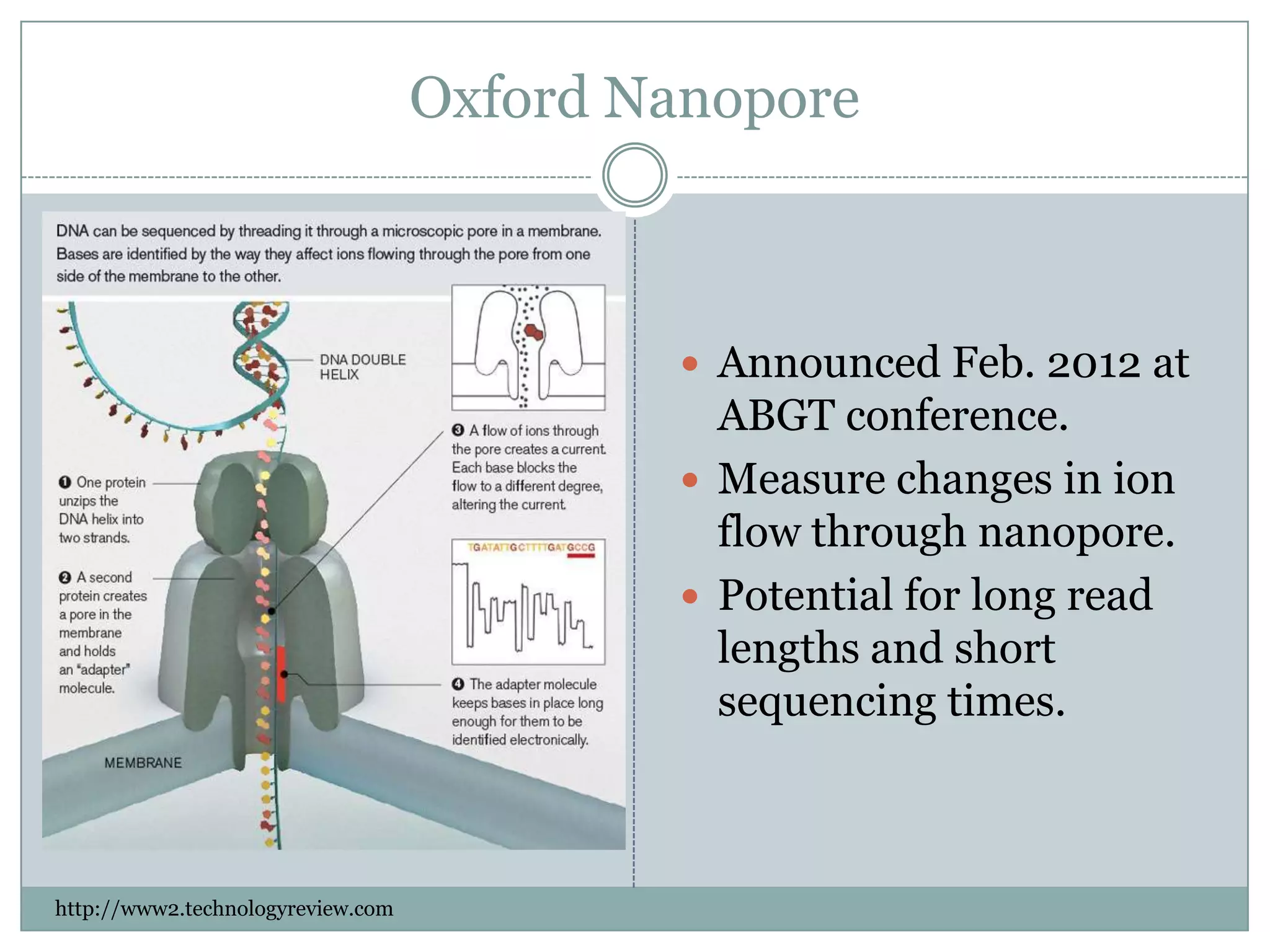
This document provides an overview and comparison of popular next-generation sequencing platforms. It discusses the common sequencing pipeline including library preparation, massively parallel sequencing, and bioinformatics analysis. Popular platforms like Roche 454, Illumina, and SOLiD are described in detail focusing on their specific sequencing chemistries and performance characteristics. Newer third-generation platforms such as Ion Torrent, PacBio, and Oxford Nanopore are also introduced. A wide range of NGS applications from whole genome sequencing to RNA-seq are outlined.