
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Monosomy 7: A Concise Guide to Causes, Symptoms, Diagnosis and Management
Similar to Monosomy 7: A Concise Guide to Causes, Symptoms, Diagnosis and Management (20)
More from Sawsan Monir
Recently uploaded
Recently uploaded (20)
Monosomy 7: A Concise Guide to Causes, Symptoms, Diagnosis and Management
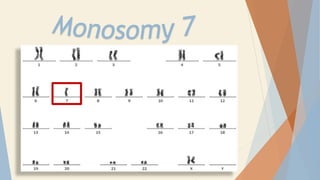
- 2. Monosomy 7 Monosomy 7 or partial deletion of the long arm of chromosome 7 (7q-) is a frequent cytogenetic finding in the bone marrow of patients with myelodysplasia (MDS) and acute myelogenous leukemia (AML). Furthermore, monosomy 7 or 7q- is the most frequent abnormality of karyotype in cases of AML that occur after cytotoxic cancer therapy or occupational exposure to mutagens. The age distribution of de novo cases shows peaks in the first and fifth decades. Monosomy 7 is found in about 5% of de novo and 40% of secondary cases of AML. These findings suggest that loss of certain genes at this region is an important event in the development of myelodysplasia
- 3. Causes In individuals with Chromosome 7, there is deletion (monosomy) of a portion of the short arm (p) of chromosome 7. In most cases, Chromosome 7, Partial Monosomy 7p appears to be caused by spontaneous (de novo) errors during early embryonic development that occur for unknown reasons. In such instances, the parents of the affected child usually have normal chromosomes and a relatively low risk of having another child with the chromosomal abnormality.
- 4. Causes Rare cases have also been reported that appear to result from balanced chromosomal rearrangements in one of the parents. Such a chromosomal rearrangement may be associated with an increased risk of abnormal chromosomal development in the carrier’s offspring. Chromosomal analysis and genetic counseling are typically recommended for parents of an affected child to help confirm or exclude the presence of a balanced chromosomal rearrangement involving chromosome 7 in one of the parents.
- 5. Signs & symptoms Patients can be asymptomatic or, if anemia is more severe, can have pallor, weakness, loss of a sense of well being, and exertional dyspnea. A small proportion of patients have infections related to severe neutropenia or neutrophil dysfunction, or hemorrhage related to severe thrombocytopenia or platelet dysfunction at the time of diagnosis. Patients with severe depressions of neutrophil and platelet counts at diagnosis usually have oligoblastic myelogenous leukemia. Rarely, patients have fever unrelated to infection. Arthralgia is the initial complaint in some patients. The presentation, infrequently, can mimic a connective tissue disease. Hepatomegaly or splenomegaly occurs in approximately 5 or 10 percent of patients, respectively.
- 6. Special Clinical Features of Myelodysplastic Syndromes Diabetes Insipidus of Myelodysplastic Syndromes Patients with an indolent phase (oligoblastic myelogenous leukemia) prior to overt AML may develop diabetes insipidus. Hypothalamic involvement can lead to polyuria, polydipsia, and decreased libido. Hypothalamic-posterior hypophysis insufficiency in clonal myeloid states is associated with monosomy 7 in hematopoietic cells. Neutrophilic Dermatosis of Myelodysplastic Syndromes Acute neutrophilic dermatosis (Sweet disease) is an acute febrile illness characterized by erythematous patches on the arms, face, and legs that progress to painful brown plaques. The plaques may ulcerate and produce large necrotizing skin lesions.
- 7. Special Clinical Features of Myelodysplastic Syndromes Inflammatory Syndromes of Myelodysplastic Syndromes Immune or inflammatory syndromes may be seen in as many as 10 percent of patients. A symptom complex that mimics systemic lupus erythematosus; fever, pleurisy, symmetric arthritis, plasma antinuclear antibody, and pancytopenia. Behçet disease, glomerulonephritis, seronegative arthritis, systemic vasculitis, polychondritis, polyneuropathy, panniculitis, and inflammatory bowel disease also have been associated with clonal myeloid disorders. Other Cancers of Myelodysplastic Syndromes The incidence of other cancers may be higher in subjects with myelodysplastic diseases.
- 8. Diagnosis the diagnosis of Chromosome 7, Partial Monosomy 7p may be suggested before birth (prenatally) by specialized tests such as ultrasound, amniocentesis, and/or chorionic villus sampling (CVS). During fetal ultrasonography, reflected sound waves create an image of the developing fetus, potentially revealing certain characteristic findings that suggest a chromosomal disorder or other abnormalities. With amniocentesis, a sample of fluid that surrounds the developing fetus is removed and analyzed, while CVS involves the removal of tissue samples from a portion of the placenta. Chromosomal analysis performed on such fluid or tissue samples may reveal the presence of Partial Monosomy 7p.
- 9. Diagnosis The syndrome may be diagnosed and/or confirmed after birth (postnatally) by a thorough clinical evaluation, identification of characteristic physical findings, and chromosomal analysis. Diagnostic evaluation may include various studies, including advanced imaging techniques, to help detect and/or characterize certain abnormalities that may be associated with the syndrome (e.g., particular craniofacial defects, musculoskeletal abnormalities, etc.). In addition, a thorough cardiac evaluation may be advised to detect any heart abnormalities that may be present. Such evaluation may include a thorough clinical examination, evaluation of heart and lung sounds through use of a stethoscope. and specialized tests that enable physicians to evaluate the structure and function of the heart (e.g., x-ray studies, electrocardiography [EKG], echocardiography).
- 10. Management Definitive therapy is bone marrow transplantation (BMT) prior to the emergence of a leukemic clone. The suitability of sibs who are potential bone marrow donors may be evaluated with appropriate hematologic and cytogenetic studies to rule out bone marrow disease associated with familial monosomy 7. However, given that the underlying germline pathogenic variant may not be known, a matched sib donor may not be an ideal candidate (unless much older than the affected individual and with no evidence of hematologic disorders ) An unrelated donor may be more suitable
- 11. Prevention of Secondary Complications Individuals with monosomy 7 have increased sensitivity to chemotherapy and radiation doses used in conventional ablative BMT approaches, and therefore require reduction in conditioning intensity.
- 12. Molecular Genetics Heterozygous microdeletion involved 3 contiguous genes, SAMD9 (sterile alpha motif domain containing 9) , SAMD9L (sterile alpha motif domain containing 9 like) , and HEPACAM2 . o These 3 genes deleted at high frequency in both adult and childhood myeloid leukemia.
- 13. Molecular Genetics Heterozygous acquired deletions at EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) and CUL1 (Cullin 1) genes in bone marrow cells o The findings suggested that EZH2 may act as a tumor suppressor gene in some cases, and likely influences epigenetic modifications that may lead to cancer, since EZH2 functions as a histone methyltransferase.
- 14. Mode of Inheritance The mode of inheritance of familial monosomy 7 is unclear. Because parents of these probands do not appear to be affected, autosomal recessive inheritance has been suggested . However, there is no report of relationships between the fathers of these kindreds, making autosomal recessive inheritance less likely.
- 15. Mode of Inheritance In one kindred, eight of 14 first cousins (the offspring of 3 sisters) developed aplastic anemia or acute myeloid leukemia (AML). Thus, in this family approximately 50% of maternal first cousins inherited a trait that resulted in aplastic anemia or AML with frequent loss of chromosome 7. Another kindred with five maternally related first cousins from two sibships shows a similar pattern of inheritance. In both of these kindreds, male and female cousins were affected, suggesting that this is not an X-linked trait.
- 16. Related Genetic Counseling Issues Family planning o The optimal time for estimating genetic risk is before pregnancy. o It is appropriate to offer genetic counseling to young adults who are affected or at risk. DNA banking is the storage of DNA (extracted from WBCs) for possible future use. o Because it is likely that testing methodology (e.g. genome sequencing) and our understanding of genes, allelic variants, and diseases will improve in the future o Consideration should be given to banking DNA of affected individual and their family members.