Download as PDF, PPTX



































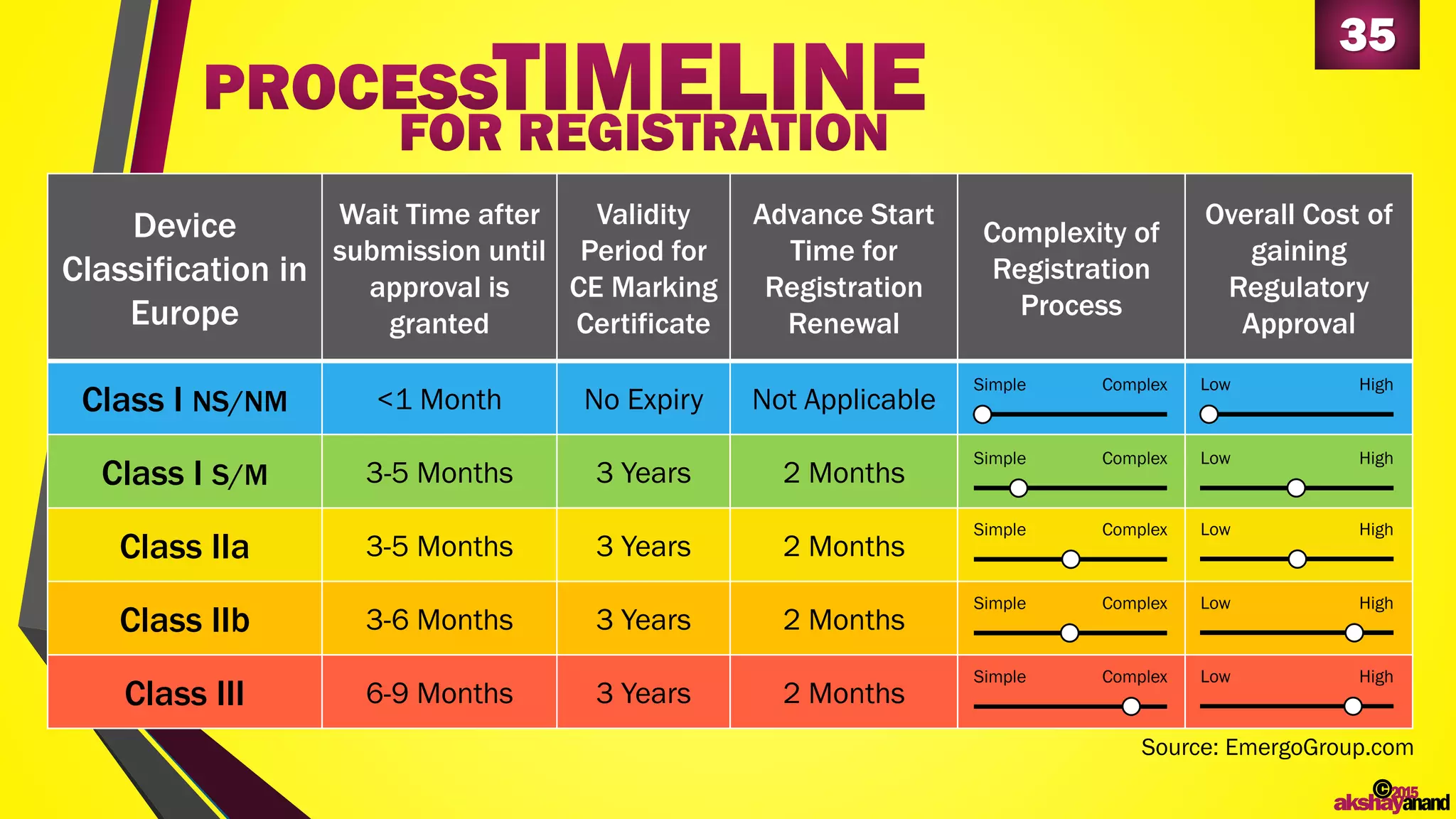




This document provides an overview of the regulatory approval process for medical devices in the European Union. It discusses key definitions such as the classification of medical devices and relevant EU directives. The approval process involves implementing quality systems, preparing a technical file or design dossier, appointing a notified body for audits and certification, registering the device, and affixing the CE mark. Vigilance reporting and post-market surveillance are also required ongoing responsibilities for device manufacturers.












![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)



































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)






