Downloaded 17 times



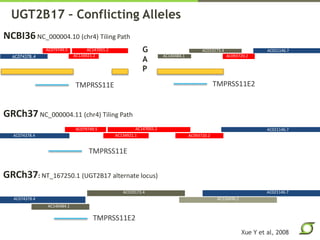
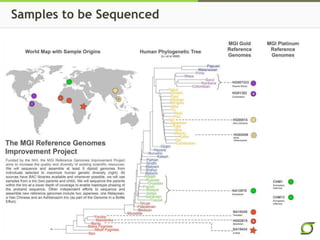
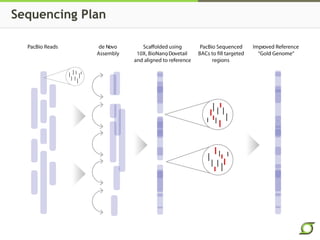
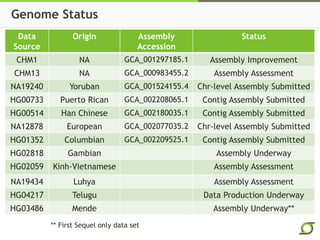
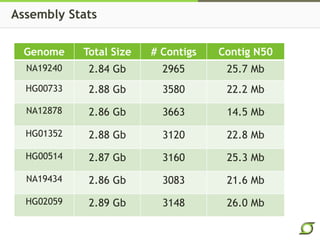
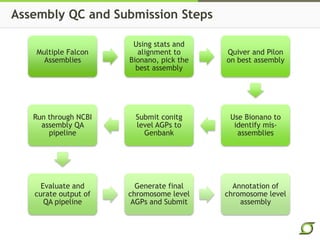
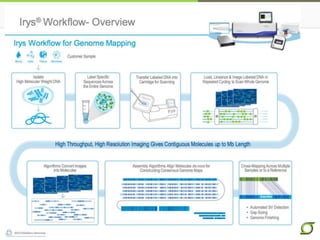
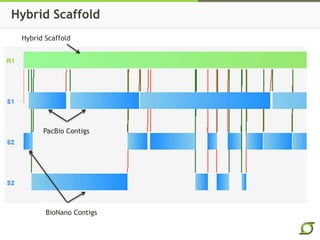
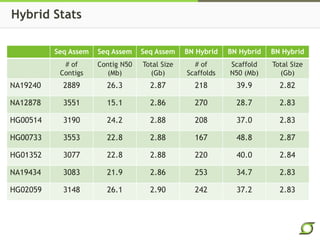
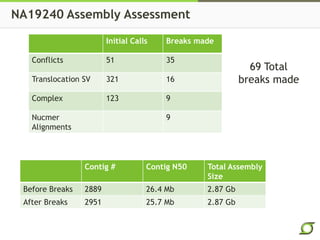
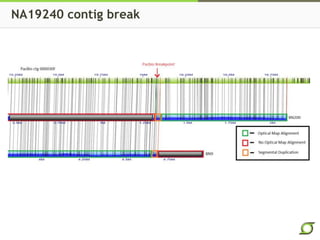
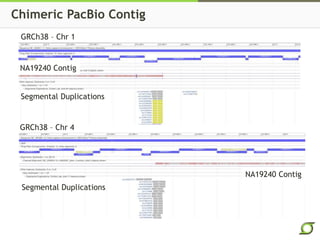
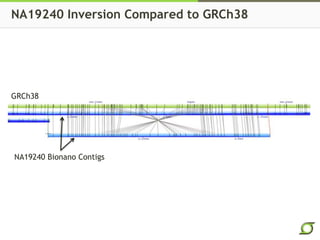
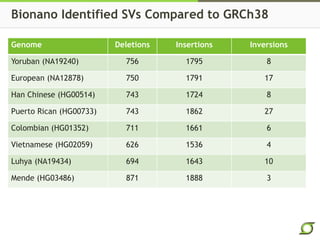
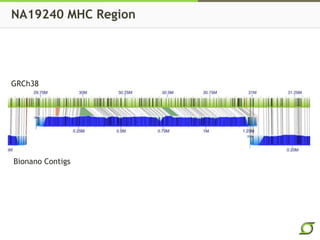
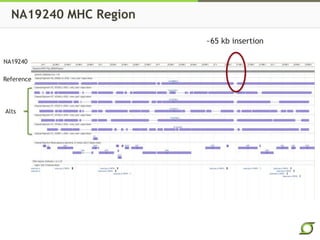
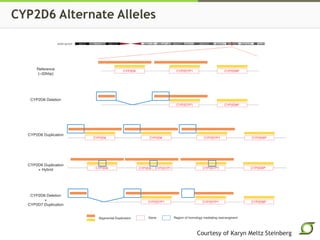
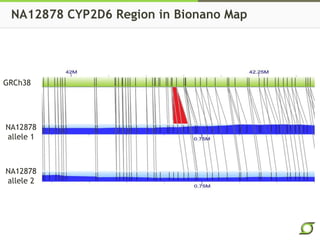
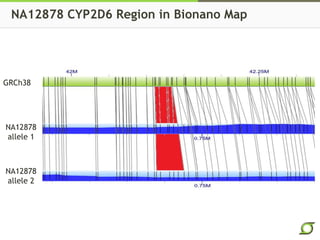

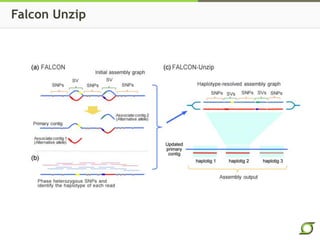
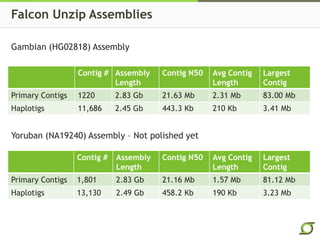
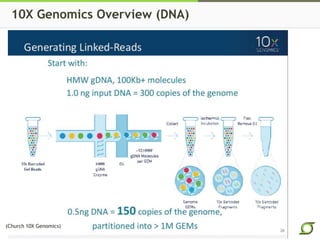




The document discusses the ongoing efforts to improve the human reference genome assembly, currently represented by GRCh38, highlighting its limitations in reflecting genetic diversity across different individuals and ancestries. It emphasizes the advancements in technology that aid genome assembly and the necessity for high-quality reference sequences to better capture human genetic variation. The document outlines plans for future assemblies, data analysis, and potential methodologies to enhance genome representation and mapping strategies.




































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)












