
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Amino acid degradation 1
Similar to Amino acid degradation 1 (20)
More from Faijanur Siddiquee
More from Faijanur Siddiquee (6)
Recently uploaded
Recently uploaded (20)
Amino acid degradation 1
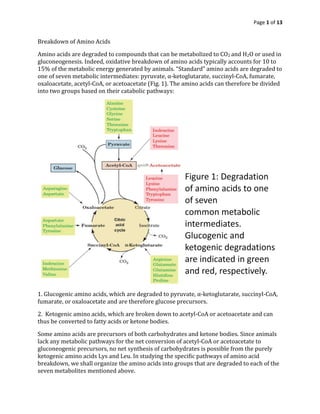
- 1. Page 1 of 13 Breakdown of Amino Acids Amino acids are degraded to compounds that can be metabolized to CO2 and H2O or used in gluconeogenesis. Indeed, oxidative breakdown of amino acids typically accounts for 10 to 15% of the metabolic energy generated by animals. “Standard” amino acids are degraded to one of seven metabolic intermediates: pyruvate, α-ketoglutarate, succinyl-CoA, fumarate, oxaloacetate, acetyl-CoA, or acetoacetate (Fig. 1). The amino acids can therefore be divided into two groups based on their catabolic pathways: 1. Glucogenic amino acids, which are degraded to pyruvate, α-ketoglutarate, succinyl-CoA, fumarate, or oxaloacetate and are therefore glucose precursors. 2. Ketogenic amino acids, which are broken down to acetyl-CoA or acetoacetate and can thus be converted to fatty acids or ketone bodies. Some amino acids are precursors of both carbohydrates and ketone bodies. Since animals lack any metabolic pathways for the net conversion of acetyl-CoA or acetoacetate to gluconeogenic precursors, no net synthesis of carbohydrates is possible from the purely ketogenic amino acids Lys and Leu. In studying the specific pathways of amino acid breakdown, we shall organize the amino acids into groups that are degraded to each of the seven metabolites mentioned above.
- 2. Page 2 of 13 Alanine, Cysteine, Glycine, Serine, and Threonine Are Degraded to Pyruvate Five amino acids—alanine, cysteine, glycine, serine, and threonine—are broken down to yield pyruvate (Fig. 2). Alanine is straightforwardly transaminated to pyruvate. Serine is converted to pyruvate through dehydration by serine dehydratase. This PLP-dependent enzyme, like the aminotransferases, forms a PLP–amino acid Schiff base which facilitates the removal of the amino acid’s α-hydrogen atom. In the serine dehydratase reaction, however, the Cα carbanion breaks down with the elimination of the amino acid’s Cβ OH, rather than with tautomerization, so that the substrate undergoes α,β elimination of H2O rather than deamination (Fig. 3).The product of the dehydration, the enamine aminoacrylate, tautomerizes nonenzymatically to the corresponding imine, which spontaneously hydrolyzes to pyruvate and ammonia.
- 3. Page 3 of 13 Glycine is converted to pyruvate by first being converted to serine by the enzyme serine hydroxymethyltransferase, another PLP-containing enzyme (Fig. 2, Reaction 4). This enzyme uses N5,N10-methylenetetrahydrofolate (N5,N10-methylene-THF) as a one- carbon donor. The methylene group of the THF cofactor is obtained from a second glycine in Reaction 3 of Fig. 2, which is catalyzed by the glycine cleavage system (called the glycine decarboxylase multienzyme system in plants). This enzyme is a multiprotein complex that resembles pyruvate dehydrogenase.
- 4. Page 4 of 13
- 5. Page 5 of 13 The serine dehydratase reaction entails a PLP-catalyzed elimination of water across the bond between the and carbons (step 1 ), leading eventually to the production of pyruvate (steps 2 through 4 ). In the serine hydroxymethyltransferase reaction, a PLP-stabilized carbanion on the carbon of glycine (product of step 1 ) is a key intermediate in the transfer of the methylene group (as -CH2-OH) from N5,N10-methylenetetrahydrofolate to form serine. This reaction is reversible. The glycine cleavage enzyme is a multienzyme complex, with components P, H, T, and L. The overall reaction, which is reversible, converts glycine to CO2 and NH4+ , with the second glycine carbon taken up by tetrahydrofolate to form N5,N10-methylenetetrahydrofolate The glycine cleavage system mediates the major route of glycine degradation in mammalian tissues. An inherited deficiency of the glycine cleavage system causes the disease nonketotic hyperglycinemia, which is characterized by mental retardation and accumulation of large amounts of glycine in body fluids. Threonine is both glucogenic and ketogenic since it generates both pyruvate and acetyl- CoA. Its major route of breakdown is through threonine dehydrogenase (Fig. 2, Reaction 6), producing α-amino-β-ketobutyrate, which is converted to acetyl-CoA and glycine by α- amino-β-ketobutyrate lyase (Fig. 2, Reaction 7). The glycine can be converted, through serine, to pyruvate. Serine Hydroxymethyltransferase Catalyzes PLP-Dependent Cα ___ Cβ Bond Formation and Cleavage. Threonine can also be converted directly to glycine and acetaldehyde (which is subsequently oxidized to acetyl-CoA) via Reaction 5 of Fig. 2, which breaks threonine’s Cα ___ Cβ bond. This PLP-dependent reaction is catalyzed by serine hydroxymethyl-transferase, the same enzyme that adds a hydroxymethyl group to glycine
- 6. Page 6 of 13 to produce serine (Fig. 2, Reaction 4). In the glycine serine reaction, the amino acid’s Cα___H bond is cleaved (as occurs in transamination) and a Cα ___Cβ bond is formed. In contrast, the degradation of threonine to glycine by serine hydroxymethyltransferase acts in reverse, beginning with Cα— Cβ bond cleavage: With the cleavage of any of the bonds to Cα, the PLP group delocalizes the electrons of the resulting carbanion. This feature of PLP action is the key to understanding how the same amino acid–PLP Schiff base can undergo cleavage of different bonds to Cα in different enzymes.
- 7. Page 7 of 13 Cysteine can be converted to pyruvate via several routes in which the sulfhydryl group is released as H2S, SO32- , or SCN-. HC + NH3 CH2 COO- SH HC + NH3 CH2 COO- SO2 - H2C + NH3 CH2 SO2 - Cysteine Cysteinesulfinate Hypotaurine H2C + NH3 CH2 SO3 - Taurine Pyruvate KG Glu HSO3 -Bisulfite HSO3 - + O2 + H2O SO4 2- + H2O2 + H+ Sulfite oxidase HC + NH3 CH2 COO- SH HC + NH3 CH2 COO- S Cysteine Cysteine H2O NH4 + SH + PyruvateThiocysteine HC + NH3 CH2 COO- SH KG Glu C O CH2 COO- SH B-Mercaptopyruvate SSO3 2- SO3 2- Pyruvate 3-mercaptopyruvate sulfurtransferase Thiosulfate Thiocysteine + Thiosulfate + CN- Rhodanese Cysteine + Thiocyanate
- 8. Page 8 of 13 Asparagine and Aspartate Are Degraded to Oxaloacetate Transamination of aspartate leads directly to oxaloacetate: Asparagine is also converted to oxaloacetate in this manner after its hydrolysis to aspartate by L-asparaginase: Interestingly, L-asparaginase is an effective chemotherapeutic agent in the treatment of cancers that must obtain asparagine from the blood, particularly acute lymphoblastic leukemia. Arginine, Glutamate, Glutamine, Histidine, and Proline Are Degraded to α-Ketoglutarate Arginine, glutamine, histidine, and proline are all degraded by conversion to glutamate , which in turn is oxidized to α-ketoglutarate by glutamate dehydrogenase. Conversion of glutamine to glutamate involves only one reaction: hydrolysis by glutaminase. In the kidney, the action of glutaminase produces ammonia, which combines with a proton to form the ammonium ion (NH4 + ) and is excreted. During metabolic acidosis, kidney glutaminase helps eliminate excess acid. Although free NH3 in the blood could serve the same acid-absorbing purpose, ammonia is toxic. It is therefore converted to glutamine by glutamine synthetase in the liver. Glutamine therefore acts as an ammonia transport system between the liver, where much of it is synthesized, and the kidneys, where it is hydrolyzed by glutaminase.
- 9. Page 9 of 13 Histidine’s conversion to glutamate is more complicated: It is nonoxidatively deaminated, then it is hydrated, and its imidazole ring is cleaved to form N-formiminoglutamate. The formimino group is then transferred to tetrahydrofolate, forming glutamate and N5 -formimino- tetrahydrofolate. Both arginine and proline are converted to glutamate through the intermediate formation of glutamate-5-semialdehyde. Isoleucine, Methionine, and Valine Are Degraded to Succinyl-CoA Isoleucine, methionine, and valine have complex degradative pathways that all yield propionyl- CoA, which is also a product of odd-chain fatty acid degradation. Propionyl-CoA is converted to succinyl-CoA by a series of reactions requiring biotin and coenzyme B12). Methionine Breakdown Involves Synthesis of S-Adenosylmethionine and Cysteine. Methionine degradation (Fig.) begins with its reaction with ATP to form S-adenosylmethionine (SAM; alternatively AdoMet). This sulfonium ion’s highly reactive methyl group makes it an important biological methylating agent. For instance, SAM is the methyl donor in the synthesis of phosphatidylcholine from phosphatidylethanolamine. Donation of a methyl group from SAM leaves S-adenosylhomocysteine, which is then hydrolyzed to adenosine and homocysteine. The homocysteine can be methylated to re-form methionine via a reaction in which N5-methyltetrahydrofolate is the methyl donor. Alternatively, the homocysteine can combine with serine to yield cystathionine, which subsequently forms cysteine (cysteine biosynthesis) and
- 10. Page 10 of 13 α-ketobutyrate. The α-ketobutyrate continues along the degradative pathway to propionyl-CoA and then succinyl-CoA. High homocysteine levels are associated with disease. Homocysteine, a Marker of Disease The cellular level of homocysteine depends on its rate of synthesis through methylation reactions utilizing SAM (Fig., Reactions 2 and 3) and its rate of utilization through remethylation to form methionine (Fig., Reaction 4) and reaction with serine to form cystathionine in the cysteine biosynthetic pathway (Fig., Reaction 5). An increase in homocysteine levels leads to hyperhomocysteinemia, elevated concentrations of homocysteine in the blood, which is associated with cardiovascular disease. The link was first discovered in individuals with homocysteinuria, a disorder in which excess homocysteine is excreted in the urine. These individuals develop atherosclerosis as children, possibly because homocysteine causes oxidative damage to the walls of blood vessels even in the absence of elevated LDL levels. Hyperhomocysteinemia is also associated with neural tube defects, the cause of a variety of severe birth defects including spina bifida (defects in the spinal column that often result in paralysis) and anencephaly (the invariably fatal failure of the brain to develop, which is the leading cause of infant death due to congenital anomalies). Hyperhomocysteinemia is readily controlled by ingesting the vitamin precursors of the coenzymes that participate in homocysteine breakdown, namely, B6 (pyridoxine, the PLP precursor), B12 , and folate. Folate, especially, alleviates hyperhomocysteinemia; its
- 11. Page 11 of 13 administration to pregnant women dramatically reduces the incidence of neural tube defects in newborns. Because neural tube development is one of the earliest steps of embryogenesis, women of childbearing age are encouraged to consume adequate amounts of folate even before they become pregnant. Around 10% of the population is homozygous for an Ala → Val mutation in N5 ,N10 -methylene- tetrahydrofolate reductase (MTHFR), which catalyzes the conversion of N5 ,N10 -methylene- THF to N5 -methyl-THF. This reaction generates the N5 -methyl-THF required to convert homocysteine to methionine (Fig. 21-18. The mutation does not affect the enzyme’s reaction kinetics but instead increases the rate at which its essential flavin cofactor dissociates. Folate derivatives that bind to the enzyme decrease the rate of flavin loss, thus increasing the enzyme’s overall activity and decreasing the homocysteine concentration. The prevalence of the MTHFR mutation in the human population suggests that it has (or once had) some selective advantage; however, this is as yet a matter of speculation. Tetrahydrofolates Are One-Carbon Carriers. Many biosynthetic processes involve the addition of a C1 unit to a metabolic precursor. In most carboxylation reactions (e.g., pyruvate carboxylase), the enzyme uses a biotin cofactor. In some reactions, S-adenosylmethionine functions as a methylating agent. However, tetrahydrofolate (THF) is more versatile than either of those cofactors because it can transfer C1 units in several oxidation states. THF is a 6-methylpterin derivative linked in sequence to a p-aminobenzoic acid and a Glu residue: Up to five additional Glu residues are linked to the first glutamate via isopeptide bonds to form a polyglutamyl tail. THF is derived from the vitamin folic acid (Latin: folium, leaf), a doubly oxidized form of THF that must be enzymatically reduced before it becomes an active coenzyme. Both reductions are catalyzed by dihydrofolate reductase (DHFR).
- 12. Page 12 of 13 Mammals cannot synthesize folic acid, so it must be provided in the diet or by intestinal microorganisms. C1 units are covalently attached to THF at positions N5, N10, or both N5 and N10. These C1 units, which may be at the oxidation levels of formate, formaldehyde, or methanol (Table), are all interconvertible by enzymatic redox reactions. THF acquires C1 units in the conversion of serine to glycine by serine hydroxymethyl- transferase, in the cleavage of glycine, and in histidine breakdown. The C1 units carried by THF are used in the synthesis of thymine nucleotides and in the synthesis of methionine from homocysteine. By promoting the latter process, supplemental folate helps prevent diseases associated with abnormally high levels of homocysteine.
- 13. Page 13 of 13 Sulfonamides (sulfa drugs) such as sulfanilamide are antibiotics that are structural analogs of the p-aminobenzoic acid constituent of THF: They competitively inhibit bacterial synthesis of THF at the paminobenzoic acid incorporation step, thereby blocking THF-requiring reactions.The inability of mammals to synthesize folic acid leaves them unaffected by sulfonamides, which accounts for the medical utility of these widely used antibacterial agents.