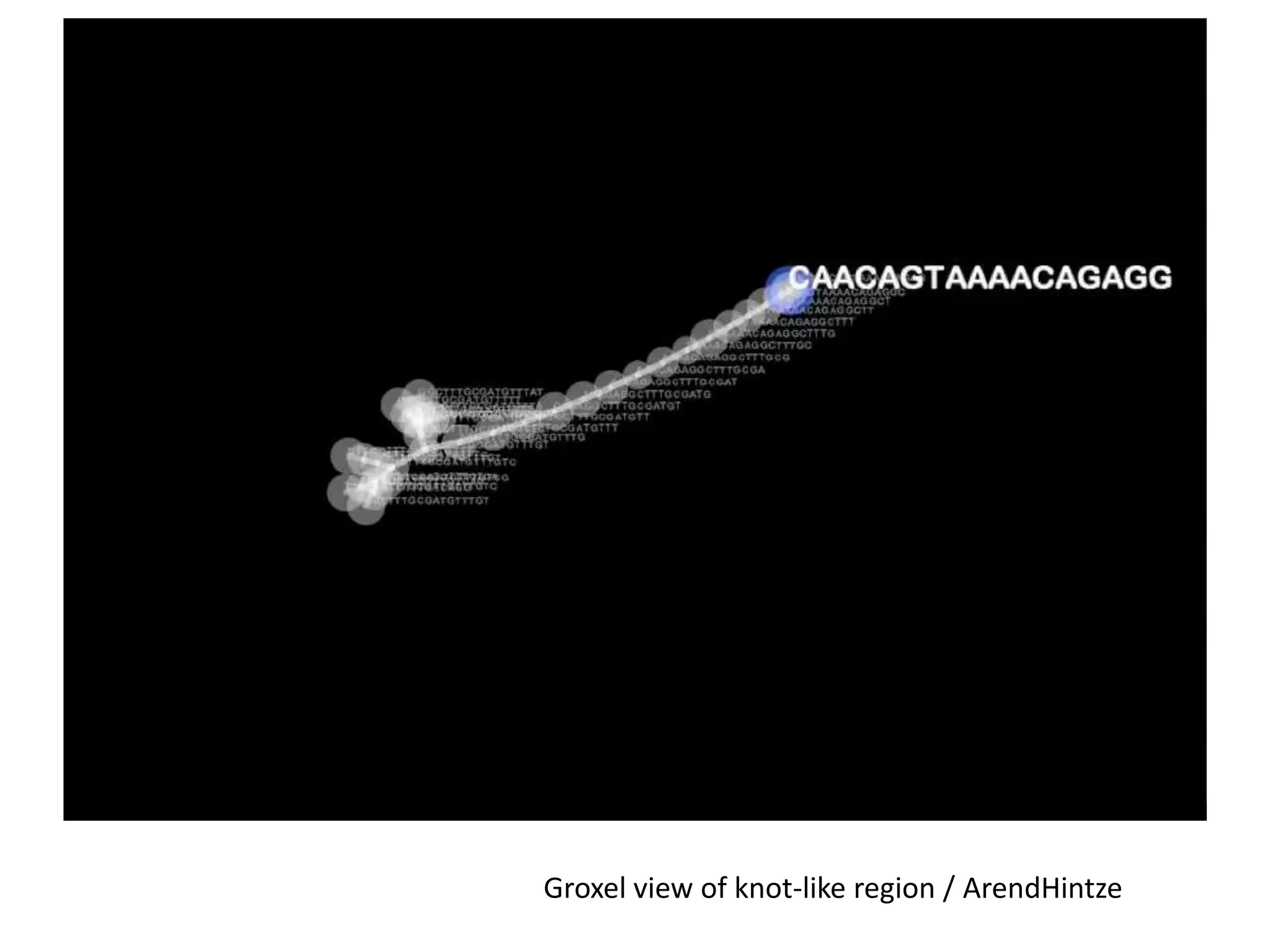
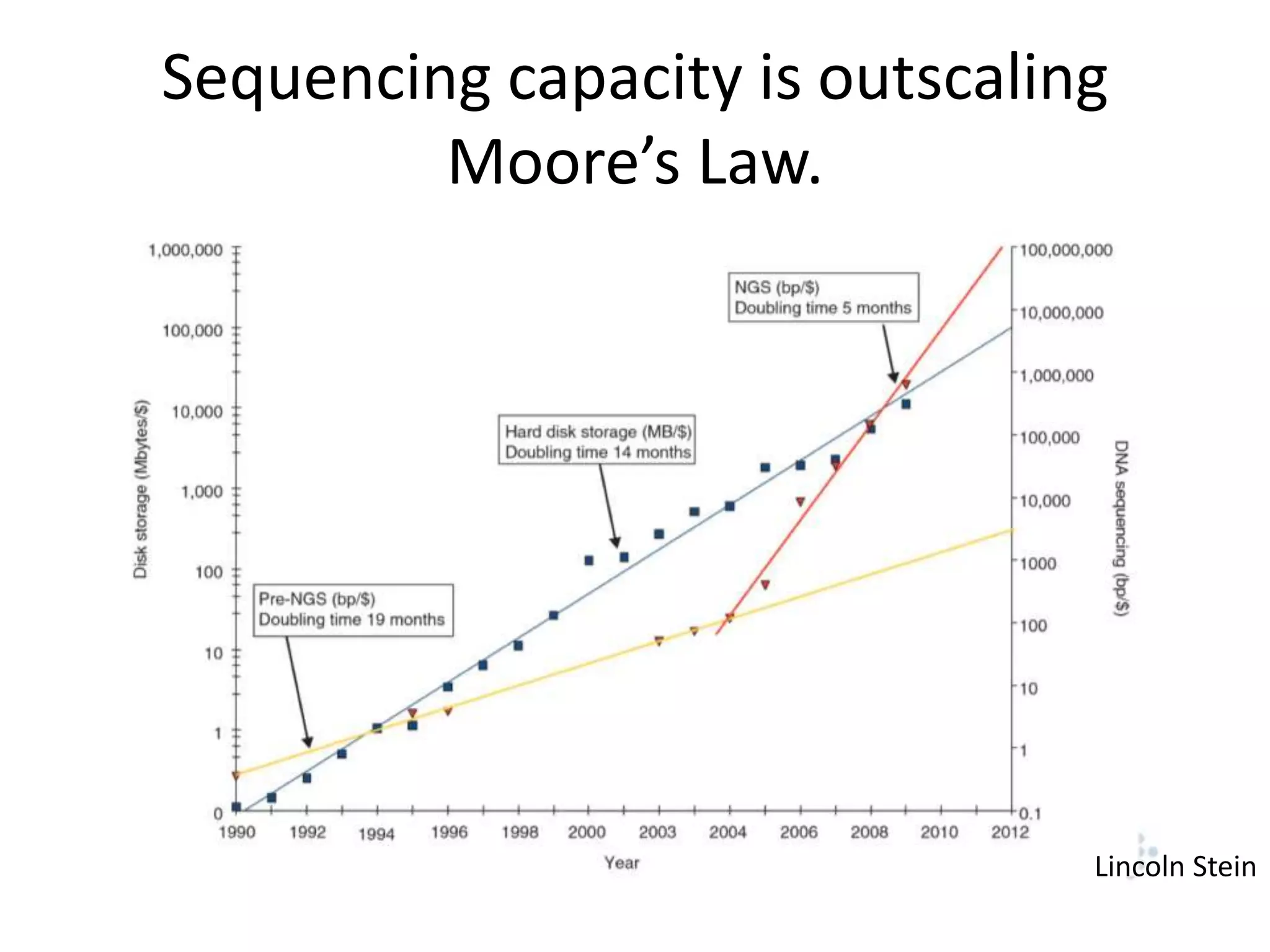
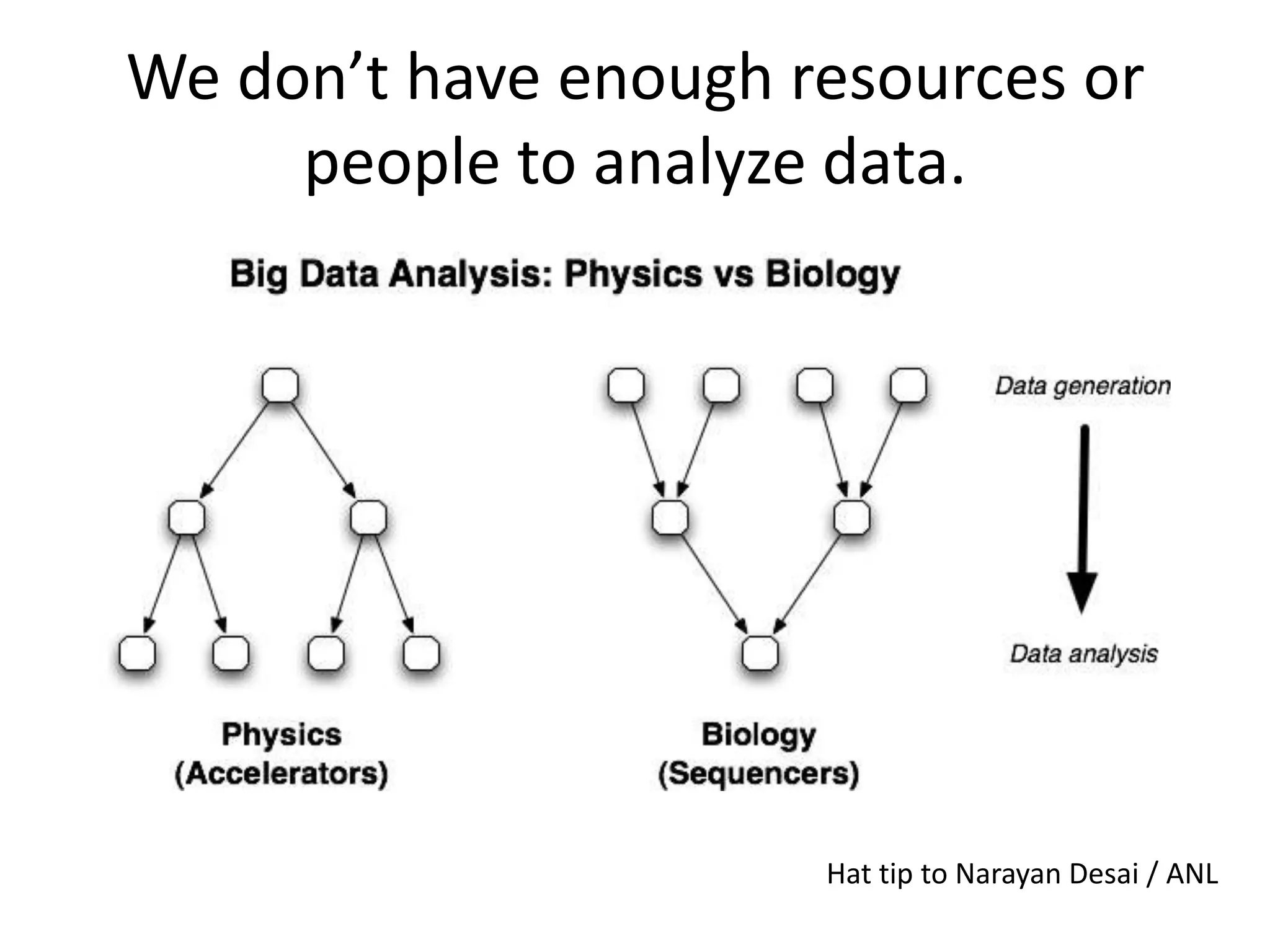
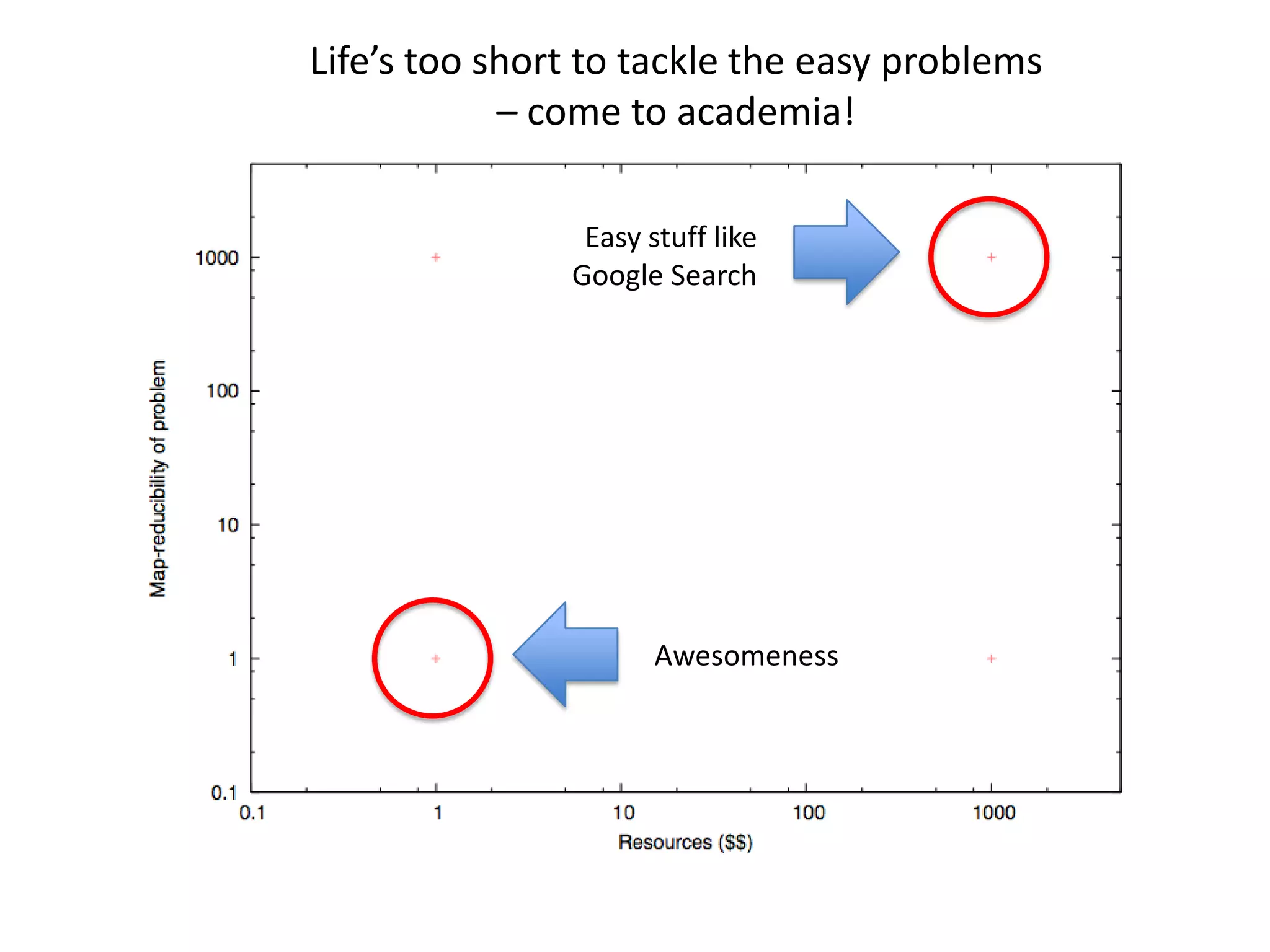
This document summarizes a talk about using probabilistic data structures like Bloom filters to handle large genomic and sequencing datasets. Bloom filters allow storing and querying enormous numbers of DNA fragments and sequences in a way that is memory efficient and scales to very large datasets. The talk describes how Bloom filters can be used to assemble genomes and reduce complexity in assembly graphs. While not perfect representations, Bloom filters enable genomic assembly and analysis that would otherwise not be possible given the volume of data.







![class BloomFilter(object): def __init__(self, tablesizes, k=DEFAULT_K):self.tables = [ (size, [0] * size) \ for size in tablesizes ]self.k = k def add(self, word): # insert; ignore collisionsval = hash(word) for size, ht in self.tables:ht[val % size] = 1 def __contains__(self, word):val = hash(word) return all( ht[val % size] \ for (size, ht) in self.tables )](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-11-2048.jpg)
![class BloomFilter(object): def __init__(self, tablesizes, k=DEFAULT_K):self.tables = [ (size, [0] * size) \ for size in tablesizes ]self.k = k def add(self, word): # insert; ignore collisionsval = hash(word) for size, ht in self.tables:ht[val % size] = 1 def __contains__(self, word):val = hash(word) return all( ht[val % size] \ for (size, ht) in self.tables )](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-12-2048.jpg)
![class BloomFilter(object): def __init__(self, tablesizes, k=DEFAULT_K):self.tables = [ (size, [0] * size) \ for size in tablesizes ]self.k = k def add(self, word): # insert; ignore collisionsval = hash(word) for size, ht in self.tables:ht[val % size] = 1 def __contains__(self, word):val = hash(word) return all( ht[val % size] \ for (size, ht) in self.tables )](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-13-2048.jpg)
![Storing words in a Bloom filter>>> x = BloomFilter([1001, 1003, 1005])>>> 'oogaboog' in xFalse>>> x.add('oogaboog')>>> 'oogaboog' in xTrue>>> x = BloomFilter([2]) >>> x.add('a')>>> 'a' in x # no false negativesTrue>>> 'b' in xFalse>>> 'c' in x # …but false positivesTrue](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-14-2048.jpg)
![Storing words in a Bloom filter>>> x = BloomFilter([1001, 1003, 1005])>>> 'oogaboog' in xFalse>>> x.add('oogaboog')>>> 'oogaboog' in xTrue>>> x = BloomFilter([2]) # …false positives>>> x.add('a')>>> 'a' in xTrue>>> 'b' in xFalse>>> 'c' in xTrue](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-15-2048.jpg)
![Storing text in a Bloom filterclass BloomFilter(object): … def insert_text(self, text): for i in range(len(text)-self.k+1):self.add(text[i:i+self.k])](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-16-2048.jpg)
![def next_words(bf, word): # try all 1-ch extensions prefix = word[1:] for ch in bf.allchars: word = prefix + ch if word in bf: yield ch# descend into all successive 1-ch extensionsdef retrieve_all_sentences(bf, start): word = start[-bf.k:]n = -1 for n, ch in enumerate(next_words(bf, word)):ss = retrieve_all_sentences(bf,start + ch) for sentence in ss: yield sentence if n < 0: yield start](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-17-2048.jpg)
![def next_words(bf, word): # try all 1-ch extensions prefix = word[1:] for ch in bf.allchars: word = prefix + ch if word in bf: yield ch# descend into all successive 1-ch extensionsdef retrieve_all_sentences(bf, start): word = start[-bf.k:]n = -1 for n, ch in enumerate(next_words(bf, word)):ss = retrieve_all_sentences(bf,start + ch) for sentence in ss: yield sentence if n < 0: yield start](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-18-2048.jpg)
![Storing and retrieving text>>> x = BloomFilter([1001, 1003, 1005, 1007])>>> x.insert_text('foo bar bazbif zap!')>>> x.insert_text('the quick brown fox jumped over the lazy dog')>>> print retrieve_first_sentence(x, 'foo bar ')foo bar bazbif zap!>>> print retrieve_first_sentence(x, 'the quic')the quick brown fox jumped over the lazy dog](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-19-2048.jpg)
![Sequence assembly>>> x = BloomFilter([1001, 1003, 1005, 1007])>>> x.insert_text('the quick brown fox jumped ')>>> x.insert_text('jumped over the lazy dog')>>> retrieve_first_sentence(x, 'the quic')the quick brown fox jumpedover the lazy dog(This is known as the de Bruin graph approach to assembly; c.f. Velvet, ABySS, SOAPdenovo)](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-20-2048.jpg)
![Repetitive strings are the devil>>> x = BloomFilter([1001, 1003, 1005, 1007])>>> x.insert_text('nanana, batman!')>>> x.insert_text('my chemical romance: nanana')>>> retrieve_first_sentence(x, "my chemical")'my chemical romance: nanana, batman!'](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-21-2048.jpg)
![Note, it’s a probabilistic data structureRetrieval errors:>>> x = BloomFilter([1001, 1003]) # small Bloom filter…>>> x.insert_text('the quick brown fox jumped over the lazy dog’)>>> retrieve_first_sentence(x, 'the quic'),('the quick brY',)](https://image.slidesharecdn.com/pycon-2011-assembly-110313084617-phpapp02/75/PyCon-2011-talk-ngram-assembly-with-Bloom-filters-22-2048.jpg)