A sterility testmay be defined as - ‘a test that
critically assesses whether a sterilized
pharmaceutical product is free from
contaminating microorganisms.
According to Indian Pharmacopeia (1996) sterility
testings are intended for detecting the presence
of viable forms of microorganisms in or on the
pharmacopeial preparations.
3.
This test isperformed on the end-product and is one of
the quality control tests specified for release of a batch
of sterile product.
The sterility test cannot be used to demonstrate the
sterility of the entire batch but it may assist in identifying
a nonsterile batch of product.
4.
Injections
Implants
Syringes
Opthalmic Preparations
Ointments and Creams
Bandages
Surgical Dressings & deviecs
Needles
Products which are necessary to
be sterilized
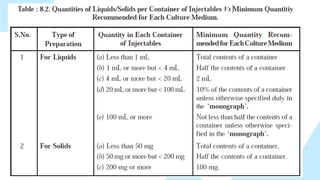
• These testsare performed based upon the principle that if
microorganisms (present in sample) are placed in a
medium which provides nutritive material and water, and
kept at a favorable temperature, the organisms will grow
and their presence can be indicated by a turbidity in the
originally clear medium.
• The interpretation of the results is based on the assumption
that the contents in every container of the batch, had they
been tested, would also have given the same results.
• Since every container can’t be tested, a sufficient number
of container should be examined to give a suitable degree
of confidence in the results of the test.
8.
Sampling Techniques
• Samplingrefers to - ‘the process of selecting a portion or
part to represent the whole’.
• In usual practice, a ‘sterility test’ attempts to infer and
ascertain the state (sterile or non-sterile) of a particular
batch; and, therefore, it designates predominantly a
‘statistical operation’.
9.
• Let usconsider that ‘p’ duly refers to the proportion of infected
containers in a batch, and ‘q’ the proportion of corresponding non-
infected containers. Then, we may have:
p + q = 1
or q = 1 – p
• Example: we may assume that a specific ‘sample’ comprising of two
items is duly withdrawn from a relatively large batch containing 10%
infected containers. Thus, the probability of a single item taken at
random contracting infection is usually given by the following
expression:
p = 0.1 [i.e., 10% = 0.1]
whereas, the probability of such an item being non-infected is invariably
represented by the following expression:
q = 1 – p = 1 – 0.1 = 0.9
10.
• Probability Status- The probability status of the said two items may be
obtained virtually in three different forms, such as :
(a) When both items get infected : p2
= 0.01
(b) When both items being non-infected :
q2
= (1 – p) 2
= (0.9) 2
= 0.81, and
(c) When one item gets infected and the other one non-infected :
1 – (p2
+ q2
)
or = 1 – (0.01 + 0.81) = 1 – (0.82)
or = 0.18
i.e., = 2pq
• Assumption : In a particular ‘sterility test’ having a ‘sample’ size of ‘n’
containers, the ensuing probability p of duly accomplishing ‘n’
consecutive ‘steriles’ is represented by the following expression:
qn
= (1 – p)n
11.
Positive Control
• Thepositive control group is a group that is designed to
produce the effect you are looking for in the experimental
group. The positive control group shows the scientists that
the desired results are possible.
• This helps prevent false negative results in the experimental
group, where a negative result is obtained but is due to a
failure in the experiment rather than a truly negative result
based on the experimental conditions.
12.
• Microorganisms forPositive Control Tests : There are,
in fact, four typical microorganisms that are being used
exclusively for the positive control tests along with their
respective type of specific enzymatic activity:
(a) Bacillus cerreus : [Broad spectrum]
(b) Staphylococcus aureus : [Penicillinase]
(c) Klebsiella aerogenes : [Penicillinase + Cephalosporinase]
(d) Enterobacter species : [Cephalosporinase]
13.
Negative Control
• Anegative control is an experimental control that does not
give a response to the test. The negative control is also not
exposed to the experimental test directly. It is done parallel
to the experiment as a control experiment.
• A negative control group serves as a benchmark to ensure
that the results that are obtained are actually due to the
independent variable and not anything else.
• An example of a negative control group is the group that
receives a placebo in clinical trials.
Prior to test,make sure that:
Media is sterile
Media supports growth of microorganisms
Media types:
1. Fluid Thioglycolate Medium
2. Fluid Soyabean-Casein Digest Medium
18.
Media Validation
MediaSterility Test:
• Negative Control - may be used to identify a “false positive”
test result.
• Incubate for 14 days prior to use, may be conducted
concurrently with test
30 - 35°C for Fluid Thioglycolate medium (FTM)
20 - 25°C for Soybean Casein Digest Medium (SCD/TSB)
• Acceptance criteria:-
Should be sterile, no growth observed
19.
Growth PromotionTest:
• To test the ability of media to support the growth of micro-
organisms.
• The media should be inoculated with <100 cfu (colony
forming unit) of challenge organisms. The challenge
inoculum should be verified by concurrent viable plate
counts.
• Growth promotion challenge organisms should show clearly
visible growth in the test media within 3 days for bacteria
and 5 days for fungi.
20.
Methods
The directinoculation:
• This method involves introducing test samples directly into nutrient
media.
• The European Pharmacopoeia recommends two media:
(1) Fluid mercaptoacetate medium (also known as fluid thioglycolate
medium), which contains glucose and sodium mercaptoacetate
(sodium thioglycolate) and is particularly suitable for the cultivation
of anaerobic organisms (incubation temperature 30-35 °C);
(2) Soyabean casein digest medium (also known as tryptone soya
broth), which will support the growth of both aerobic bacteria
(incubation temperature 30-35 °C) and fungi (incubation temperature
20-25 °C).
21.
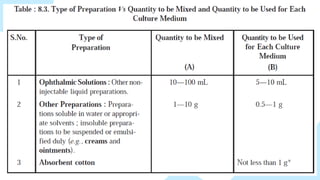
• Other mediamay be used provided that they can be shown to be
suitable alternatives.
• Limits are placed upon the ratio of the weight or volume of added
sample relative to the volume of culture medium so as to avoid reducing
the nutrient properties of the medium or creating unfavourably high
osmotic pressures within it.
22.
Membrane filtration:
•This technique is recommended by most pharmacopoeias and,
consequently, the method by which the great majority of products are
examined.
• It involves filtration of fluids through a sterile membrane filter (pore size
≤ 0.45 μm), any microorganism present being retained on the surface of
the filter.
• After washing, the filter is divided aseptically and portions are
transferred to suitable culture media which are then incubated at the
appropriate temperature for the required period of time.
• Water – soluble solids can be dissolved in a suitable diluent and
processed in this way and oil – soluble products may be dissolved in a
suitable solvent, e.g. isopropyl myristate.
23.
Another sensitivemethod for detecting low levels of
contamination in intravenous infusion fluids involves the
addition of a concentrated culture medium to the fluid in
its original container, such that the resultant mixture is
equivalent to a single strength culture medium. In this
way, sampling of the entire volume is achieved.
Pyrogens
• The agentsresponsible for this fever were termed
‘pyrogens ’.
• In theory a pyrogen is any substance that, when injected
into a mammal, elicits a rise in body temperature, and
substances produced by some bacteria, mycobacteria,
fungi and also viruses.
• Pyrogens not only increase body temperature, but also
elevate the circulating levels of inflammatory cytokines,
which may be followed by fever, blood coagulation,
hypotension, lymphopenia, neutrophilia, elevated levels of
plasma cortisol and acute - phase proteins.
26.
Sources of Pyrogens
•The sources of pyrogens in parenteral products include:
(1) water used at the end stages of the purification and crystallization of
the drug or excipients
(2) water used during processing
(3) packaging components and
(4) the chemicals, raw materials or equipment used in the preparation of
the product
(5) Incomplete removal of the microorganisms during purification of the
biologically produced drugs
27.
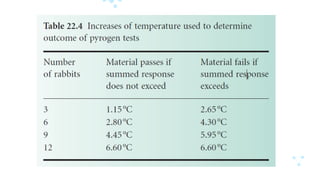
Test of Pyrogens
•Pyrogens have traditionally been assessed using rabbits
which are stored in carefully controlled conditions and
whose temperature is monitored before the administration
of the test product.
• According to BP, this test is initially based on three rabbits;
the number is progressively increased if the results fall
between the two values.
• Samples under test are injected into the marginal ear vein
at a dose no greater than 10 ml/kg.
• The animals are monitored for the 3 hour period
immediately after injection, at 30 minute intervals.
29.
• A numberof limitations of the rabbit pyrogen test are recognized:
1. Repeated use of animals leads to endotoxin tolerance.
2. There is low reactivity to the endotoxin produced by certain species,
e.g. Legionella.
3. There is also variability in control results when identical standardized
endotoxin preparations are used, which is probably related to
interlaboratory factors and variations due to seasons, rabbit species
and other biological sources.
4. Care must be taken in testing radiopharmaceuticals, and certain
drugs may themselves elicit a rise in temperature on administration.
5. This test is insufficiently sensitive to detect endotoxin in intrathecal
products where only low levels of pyrogens are acceptable.
30.
Measurement of bacterial
endotoxins
•The limulus amoebocyte lysate (LAL) test is considerably
more sensitive than the pyrogen test.
• As the Legionella endotoxin is not very pyrogenic to rabbits
it is easily detected by the LAL test.
• It has been estimated that there is a 1000 - fold difference in
sensitivity between the two tests, but the LAL test only
detects endotoxins of Gram - negative bacteria and not all
pyrogens. However, the LAL test may be used for
radiopharmaceuticals.
31.
• LAL testreagent comes from the American horseshoe crab Limulus
polyphemus.
• The endotoxin - induced coagulation of its blood is based on an enzyme
– mediated interaction of LAL with endotoxins.
• The reagents are obtained from the blood of freshly captured horseshoe
crabs whose amoebocytes are concentrated, washed and lysed with
endotoxin - free water.
• The LAL is separated from the remaining cellular debris and its activity
optimized using metallic cations, pH adjustment and additives and then
freeze - dried.
• The samples of products are incubated with LAL at 37 °C. If endotoxins
are present a solid gel forms, indicating the presence of endotoxins.
32.
Depyrogenation and theproduction
of
apyrogenic products
• Pyrogens and endotoxins are difficult to remove from products once
present and it is easier to keep components relatively endotoxin - free
rather than to remove them from the final product.
1. Rinsing or dilution is one way of eliminating pyrogenic activity
provided that the rinsing fluid is apyrogenic.
2. Closures and vials should be washed with pyrogen - free water
before sterilization.
3. Pyrogens in vials or glass components may be destroyed by dry
heat sterilization at high temperatures.
33.
4. The removalof pyrogens from Water for Injections may be effected
by distillation or reverse osmosis. Distillation is the most reliable
method for removing endotoxin.
5. Another source of endotoxins is the Water for Injection system.
Generally, circulating hot water at temperatures above 75 °C
provides an environment that is not conducive to microbial growth
and thus the formation of endotoxin.
6. Pyrogen - free water can be produced using an ultrafiltration
membrane with a nominal molecular weight limit that is low enough
to ensure the removal of endotoxins under all conditions.








![• Let us consider that ‘p’ duly refers to the proportion of infected
containers in a batch, and ‘q’ the proportion of corresponding non-
infected containers. Then, we may have:
p + q = 1
or q = 1 – p
• Example: we may assume that a specific ‘sample’ comprising of two
items is duly withdrawn from a relatively large batch containing 10%
infected containers. Thus, the probability of a single item taken at
random contracting infection is usually given by the following
expression:
p = 0.1 [i.e., 10% = 0.1]
whereas, the probability of such an item being non-infected is invariably
represented by the following expression:
q = 1 – p = 1 – 0.1 = 0.9](https://image.slidesharecdn.com/sterility-250308112828-179a1c8c/85/Sterility-Testing-for-Microbiology-pptx-9-320.jpg)


![• Microorganisms for Positive Control Tests : There are,
in fact, four typical microorganisms that are being used
exclusively for the positive control tests along with their
respective type of specific enzymatic activity:
(a) Bacillus cerreus : [Broad spectrum]
(b) Staphylococcus aureus : [Penicillinase]
(c) Klebsiella aerogenes : [Penicillinase + Cephalosporinase]
(d) Enterobacter species : [Cephalosporinase]](https://image.slidesharecdn.com/sterility-250308112828-179a1c8c/85/Sterility-Testing-for-Microbiology-pptx-12-320.jpg)