Downloaded 13 times




















![INNUENDO project [GP/EFSA/AFSCO/2015/01/CT2]
BacGenTrack project [FCT / Scientific and Technological Research Council of Turkey, TUBITAK/0004/2014]
ONEIDA project (LISBOA-01-0145-FEDER-016417) co-funded by FEEI - “Fundos Europeus Estruturais e de
Investimento” from “Programa Operacional Regional Lisboa 2020” and by national funds from FCT -
“Fundação para a Ciência e Tecnologia”
Disclaimer
The conclusions, findings, and opinions expressed in this presentation reflect only the
view of the INNUENDO consortium members and not the official position of the
European Food Safety Authority nor of the Government of the Basque Country that are
not responsible for any use that may be made of the information they contain.](https://image.slidesharecdn.com/ngsbasel2sep2017vf-170905100926/85/Software-Pipelines-The-Good-The-Bad-and-The-Ugly-34-320.jpg)

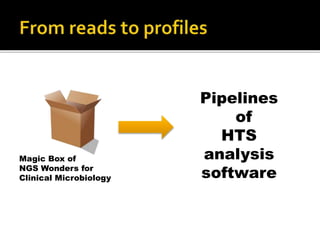
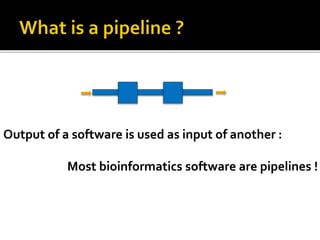
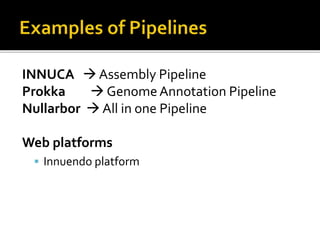
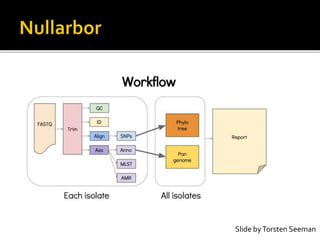
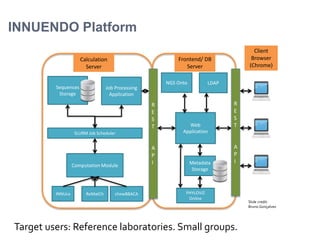
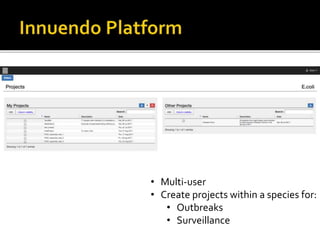
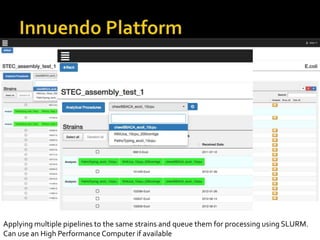
The document discusses the application of whole genome sequencing (WGS) for clinical microbiology, highlighting a software pipeline that facilitates species identification, antibiotic resistance profiling, and virulence factor analysis. It addresses the complexities of managing bioinformatics workflows, emphasizing issues such as software validation, reproducibility, and database dependencies, while also proposing solutions like the use of web platforms to simplify these processes for non-experts. Ultimately, it underscores the need for specialized personnel to maintain high-performance computing clusters essential for effective data analysis.






















































![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)












