Downloaded 18 times






















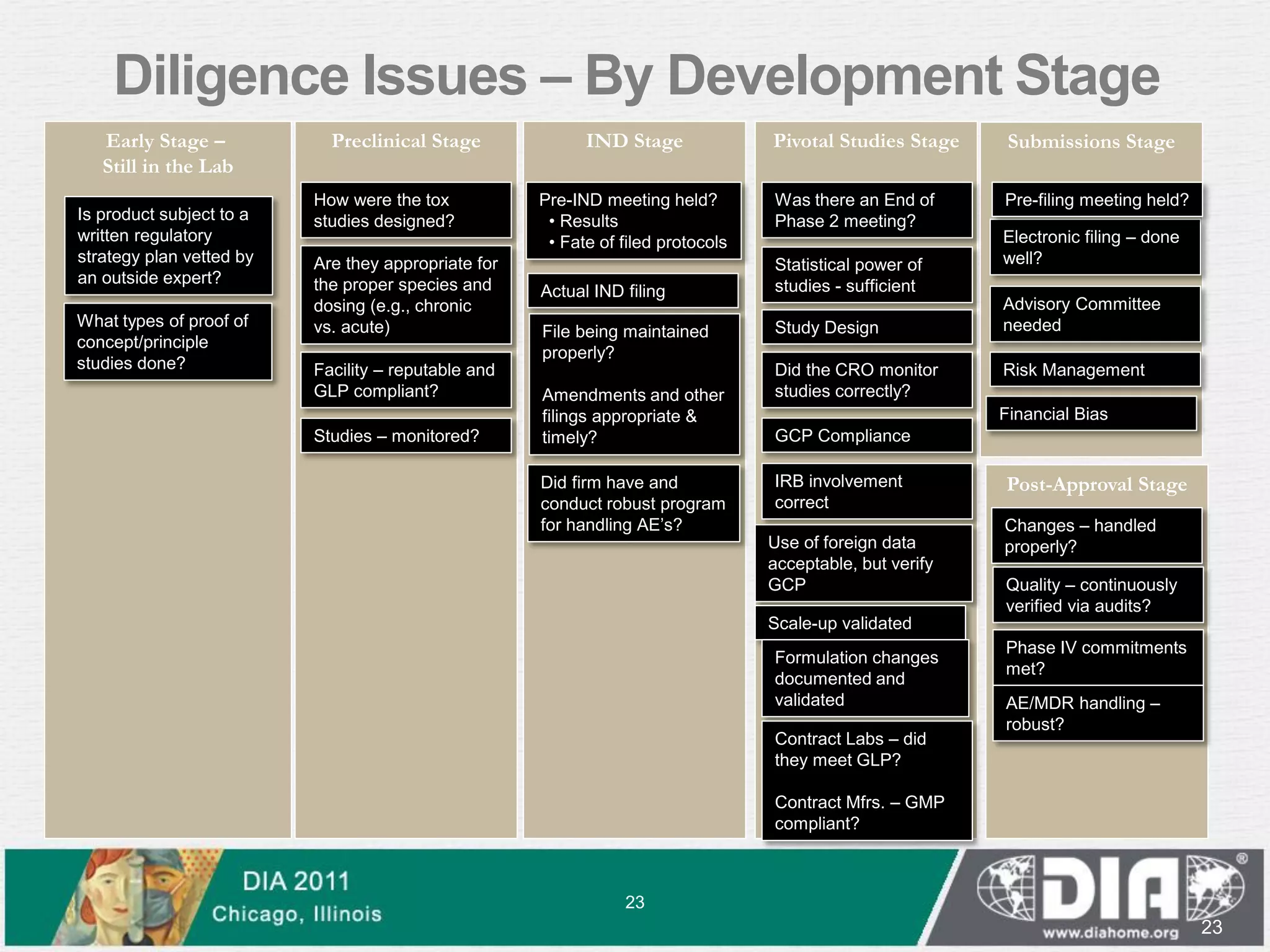




The document discusses quality considerations in due diligence for pharmaceutical transactions. It outlines general considerations for due diligence structure and challenges. It emphasizes that quality matters because drugs made in non-compliant facilities can be considered adulterated by the FDA and result in criminal and civil penalties. The document provides examples of problems to look for, including manufacturing, pharmacovigilance, compliance history, FDA inspection history, and audits. It discusses techniques for probing issues, including reviewing FDA correspondence and litigation. Special considerations are outlined for different drug development stages. Helpful charts on diligence issues by stage and analyzing identified issues are presented.























































































