Protein Secondary Structure, Ramachandran Plot, Molecular Docking, Pharmacophore, Heat Map
•
3 likes•171 views

Protein Secondary Structure, Ramachandran Plot, Molecular Docking, Pharmacophore, Heat Map explained in short. Easy to understand. Examples, FAQ given/

Recommended
More Related Content
What's hot
What's hot (20)
Similar to Protein Secondary Structure, Ramachandran Plot, Molecular Docking, Pharmacophore, Heat Map
Similar to Protein Secondary Structure, Ramachandran Plot, Molecular Docking, Pharmacophore, Heat Map (20)
More from Pradipta Banerjee
More from Pradipta Banerjee (11)
Recently uploaded
Recently uploaded (20)
Protein Secondary Structure, Ramachandran Plot, Molecular Docking, Pharmacophore, Heat Map
- 1. Pradipta Banerjee, Ph.D. Dept. of Biochemistry & Plant Physiology, CUTM, Odisha, India pradipta.banerjee@cutm.ac.in
- 2. Nomenclature of C-atoms Pradipta Banerjee, Ph.D.
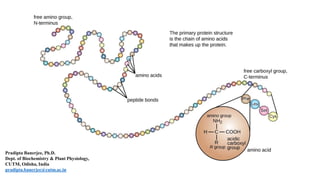
- 3. Four Levels of Organization in the Structure of a Protein The amino acid sequence is known as primary structure of protein. Stretches of polypeptide chain that form α helices and β sheets constitute protein’s secondary structure. Full three-dimensional organization of a polypeptide chain is referred as the protein’s tertiary structure, and if a particular protein molecule is formed as a complex of more than one polypeptide chain, the complete structure is designated as the quaternary structure. Pradipta Banerjee, Ph.D.
- 4. Characteristic Features 1. Dipeptide bond links 2 amino acids 2. Strong bond, Rigid, planar structure 3. Trans configuration 4. -NH and –CO are polar and can form H-bonds 5. Have 40% partial double bond character, due to resonance. Peptide Bonds Pradipta Banerjee, Ph.D.
- 5. Cis & Trans Peptide Bonds Cis: Cα groups on same side of peptide bond (very rare) Trans: Cα groups on same side of peptide bond Pradipta Banerjee, Ph.D.
- 6. Peptide groups, with few exceptions, assume the trans conformation: that in which successive Cα atoms are on opposite sides of the peptide bond joining them. This is partly a result of steric interference, which causes the cis conformation to be about 8 kJ.mol-1 less stable than the trans conformation (this energy difference is somewhat less in peptide bonds followed by a Pro residue and, in fact, 10% of the Pro residues in proteins follow a cis peptide bond, whereas cis peptides are otherwise extremely rare). …Cis & Trans Peptide Bonds Pradipta Banerjee, Ph.D.
- 7. Proline, a cyclic secondary amino acid, has conformational constraints imposed by the cyclic nature of its pyrrolidine side chain, which is unique among the “standard” 20 amino acids. Pradipta Banerjee, Ph.D.
- 8. Proteins are molecules that consist of one or more polypeptide chains. These polypeptides range in length from ~40 to ~34,000 amino acid residues (although few have more than 1500 residues) and, since the average mass of an amino acid residue is about 110 D, have molecular masses that range from ~40 to over ~3700 kD. Polypeptide Chains Pradipta Banerjee, Ph.D.
- 9. Polypeptide Backbone Conformations (of a Protein) May Be Described by their Torsion Angles Torsion angles or rotation angles or dihedral angles are formed between the Cα-N bond (Φ) and the Cα-C bond (Ψ) of each of its amino acid residues. These angles, Φ and Ψ are both defined as 180° when the polypeptide chain is in its planar, fully extended (all-trans) conformation and increase for a clockwise rotation when viewed from Cα. Pradipta Banerjee, Ph.D.
- 10. α-Helix The right-handed α-helix: Hydrogen bonds between the N-H groups and the groups that are four residues back along the polypeptide chain are indicated by dashed lines**. The left-handed α-helix has never been observed (its helical parameters are mildly forbidden) is that its side chains contact its polypeptide backbone too closely. NB: 1-2% of the individual non-Gly residues in proteins assume this conformation. The α-helix is a common secondary structural element of both fibrous and globular proteins. **Hydrogen bonds of an helix are arranged such that the peptide N-H bond of the 10th residue points along the helix toward the peptide group of the 6th residue. Pradipta Banerjee, Ph.D.
- 11. If a polypeptide chain has a long block of Glu residues, this segment of the chain will not form an helix at pH 7.0. The negatively charged carboxyl groups of adjacent Glu residues repel each other so strongly that they prevent formation of helix. For the same reason, if there are many adjacent Lys and/or Arg residues, which have positively charged R groups at pH 7.0, they will also repel each other and prevent formation of the helix. Bulk and shape of Asn, Ser, Thr, and Cys residues can also destabilize an helix if they are close together in chain. A Pro residue introduces a destabilizing kink in an helix. Proline is only rarely found within an helix. Glycine occurs infrequently in helices for a different reason: it has more conformational flexibility than other amino acid residues. Polymers of glycine tend to take up coiled structures quite different from an helix. Points to Remember: α-Helix Pradipta Banerjee, Ph.D.
- 12. Βeta pleated sheet’s conformation has repeating Φ and Ψ angles that fall in the allowed region of the Ramachandran diagram and utilizes the full hydrogen bonding capacity of the polypeptide backbone. In pleated sheets, however, hydrogen bonding occurs between neighboring polypeptide chains rather than within one as in helices. Pleated sheets come in two varieties: 1. The antiparallel pleated sheet, in which neighboring hydrogen bonded polypeptide chains run in opposite directions. 2. The parallel pleated sheet, in which the hydrogen bonded chains extend in the same direction. β-pleated sheets Pradipta Banerjee, Ph.D.
- 13. β-pleated sheets Pradipta Banerjee, Ph.D.
- 14. (a) The hairpin connection between antiparallel strands is topologically in the plane of the sheet. (b) A right-handed crossover connection between successive strands of a parallel sheet. Nearly all such crossover connections in proteins have this chirality. (c) A left-handed crossover connection between parallel sheet strands. Connections with this chirality are rare. Connections between adjacent polypeptide strands in β pleated sheets Pradipta Banerjee, Ph.D.
- 15. In the early 1960’s, G. N. Ramachandran (University of Madras, India) and co-workers computationally determined the phi and psi angles that avoid steric collisions, initially treating the atoms simply as rigid spheres. They showed that the physically allowed angle combinations (that avoid clashes) correspond largely to the secondary structures observed in proteins: alpha helices, beta sheets, and turns. Ramachandran and team also showed that the major effect of sidechains on the allowed phi and psi angles is due to Cβ. Sidechains larger than that of alanine affect the allowed angles by only a few percent. The Ramachandran Plot shows the phi and psi angles actually observed in proteins. Ramachandran Plot Pradipta Banerjee, Ph.D.
- 16. Clashes: where non-bonded atoms are overlapping, and thus in physically impossible positions. (This model simulation allows two atoms to overlap, unlike real atoms.) Peptide Bond (Pink) Plane (Yellow) of a peptide bond. Pradipta Banerjee, Ph.D.
- 17. The Ramachandran Principle says that alpha helices, beta strands, and turns are the most likely conformations for a polypeptide chain to adopt, because most other conformations are impossible due to steric collisions between atoms. Ramachandran Principle Pradipta Banerjee, Ph.D.
- 18. The conformation angles (Φ and Ψ) of several secondary structures NB: Gly, the only residue without a Cβ atom, is much less sterically hindered than the other amino acid residues. Pradipta Banerjee, Ph.D.
- 19. How many bonded atoms are required to constitute a dihedral (torsion) angle, such as phi or psi? a. None b. 1 c. 2 d. 3 e. 4 f. 5 Answer: The phi dihedral (torsion) angle is defined by: (1) the carboxy carbon from the previous amino acid; (2) N in the amino acid containing the phi bond; (3) Cα in the amino acid containing the phi bond; and (4) the carboxy carbon of the amino acid containing the phi bond. Pradipta Banerjee, Ph.D.
- 20. The number of phi and psi angles in an isolated amino acid (not in a polypeptide chain) is: a. None b. 1 c. 2 d. 3 e. 4 f. 5 Answer: The phi angle involves the carboxy carbon of the previous amino acid in a polypeptide chain. The psi angle involves the N of the subsequent amino acid. Therefore an isolated single amino acid has neither phi nor psi angles Pradipta Banerjee, Ph.D.
- 21. The number of atoms held into a geometric plane by a peptide bond is: a. None b. 1 c. 2 d. 3 e. 4 f. 5 g. 6 Pradipta Banerjee, Ph.D.
- 22. The peptide bond is unable to rotate because: a. It is a covalent bond. b. It is a non-covalent bond. c. Rotation would cause clashes. d. It is a partially double bond. Phi and psi angles directly determine a. Primary structure. b. Secondary structure. c. Tertiary structure. d. Quaternary structure Pradipta Banerjee, Ph.D.
- 23. Alpha helices are compatible with a. All possible phi-psi angle combinations. b. A limited range of phi-psi angle combinations. c. A limited range of phi angles with all possible psi angles. d. A limited range of psi angles with all possible phi angles. Pradipta Banerjee, Ph.D.
- 24. Common secondary structures are energetically favoured because a. They optimize main-chain hydrogen bonds. b. They represent all possible conformations. c. They maximize clashes between atoms. d. They minimize clashes between atoms. Overlap of van der Waals radii a. Between two non-bonded atoms is called a clash. b. Between two covalently bonded atoms is called a clash. c. Occurring between two non-bonded atoms in a molecular model signifies an energetically favourable interaction. d. Is physically impossible between two non-bonded real atoms. Pradipta Banerjee, Ph.D.
- 25. In a Ramachandran plot, the dots cluster a. Where clashes occur. b. Where clashes do not occur. c. Where alpha helices occur. d. Where beta strands occur. e. Where alpha helices, beta strands, and turns do not occur. f. Where the phi and psi angles are energetically favourable. Pradipta Banerjee, Ph.D.
- 26. Molecular docking: Predict the preferred orientation of one molecule to a second, when bound to each other to form a stable complex. Depending upon binding properties of ligand and target, it predicts the 3-D structure of any complex. Applications: 1. Drug design 2. Analysis of molecular interactions 3. Identification of role of phytochemicals in curing diseases 4. Aptamer selection • Biosensor development • Targeted drug delivery Molecular Docking Pradipta Banerjee, Ph.D.
- 27. Molecular Docking Docking can be between: • Protein - Ligand • Protein – Protein • Protein – Nucleotide The ligand acts as signal triggering molecule in case of protein-ligand binding by binding the specific site on the protein where it is targeted. Pradipta Banerjee, Ph.D.
- 28. Work Flow of Molecular Docking Pradipta Banerjee, Ph.D.
- 29. Most relevant pharmacophore features Examples hydrogen bond acceptors hydroxyl or carbonyl groups hydrogen bond donors hydroxyl or amide groups positively charged groups guanidines negatively charged groups carboxylates hydrophobic groups cyclohexyl or isopropyl groups aromatic moieties phenyl ring or other aromatic ring systems Pharmacophore Pradipta Banerjee, Ph.D. A pharmacophore is best defined as the arrangement of these functional groups in an active compound that are crucial for its activity. Pharmacophores can be derived from 2-D or 3-D molecular representations, but 3D pharmacophores are most popular (considering the fact that molecules are active in three dimensions).
- 30. Molecular Mechanics Quantum Mechanics Structure-based Drug Design or Target-based Drug Design (Docking Studies) Ligand-based Drug Design Pharmacophore Mapping Three-dimensional (3D) Structure of Target Known (Receptor/enzyme) Three-dimensional (3D) Structure of Target NOT Known/available Specify the binding site surrounding the bound co-crystal ligand Homology Modeling of Target protein / receptor Search / Identify binding site in Homology Model Specify the binding-site Docking studies Pharmacophore Modelling Pradipta Banerjee, Ph.D.
- 31. Structure Based Drug Design Pradipta Banerjee, Ph.D.
- 32. This method will Begin with biologically active compounds. Describe what chemistry those compounds have in common. Few new compounds that match this description. Compounds that match the description will also be active. Initial two steps involving the process called ‘model building’ while the final two steps are known as ‘database screening’. Ligand Based Drug Design Pradipta Banerjee, Ph.D.
- 33. Pradipta Banerjee, Ph.D. 70% hits was reduced after primary assessment 99% hits was reduced after molecular docking Case Study Stem and bark of Camptothecin acuminate Camptothecin (CPT), targets DNA Topoisomerase I In order to predict structurally diverse novel chemical entity as Top1 poison with better efficacy, Ligand-based-pharmacophore model was developed. 33% database was reduced after screening
- 34. Heat Maps In heat maps the data is displayed in a grid where each row represents a gene and each column represents a sample. The colour and intensity of the boxes is used to represent changes (not absolute values) of gene expression. Upregulated genes Downregulated genes Unchanged expression Many of the methods for visualising and interpreting gene expression data can be used for both microarray and RNA-seq experiment. HM is one of them. Pradipta Banerjee, Ph.D.
- 35. samples proteins Proteins are divided into two groups (left dendrogram) roughly corresponding to membrane-specific proteins and other proteins. Samples are divided into three groups (upper dendrogram). • The large cluster on the left corresponds to samples quantified with the URZB and URNB extraction methods. • The middle cluster includes samples quantified with the TCA1 method • The last one involves the TEAL method 1847 proteins quantified on 19 samples extracted from one maize leaf and according to 4 extraction methods based on label-free shotgun proteomics Pradipta Banerjee, Ph.D.
- 36. • Samples extracted using the URZB and URNB methods have a relatively high expression of the top cluster of proteins compared to the lower cluster. • On the other hand, the TEAL samples (right part of the map) have a relatively low for the top protein cluster and relatively high for the bottom cluster. • Finally, the TCA1 samples (middle cluster) seem to exhibit intermediate quantities for most proteins (nevertheless, proteins at the bottom of the map are relatively more abundant than proteins at the top). Low values High value Intermediate values Heat Map Analysis Pradipta Banerjee, Ph.D.
- 37. THANK YOU