Cushing’s Syndrome
Clinical Features,Diagnosis, and Management
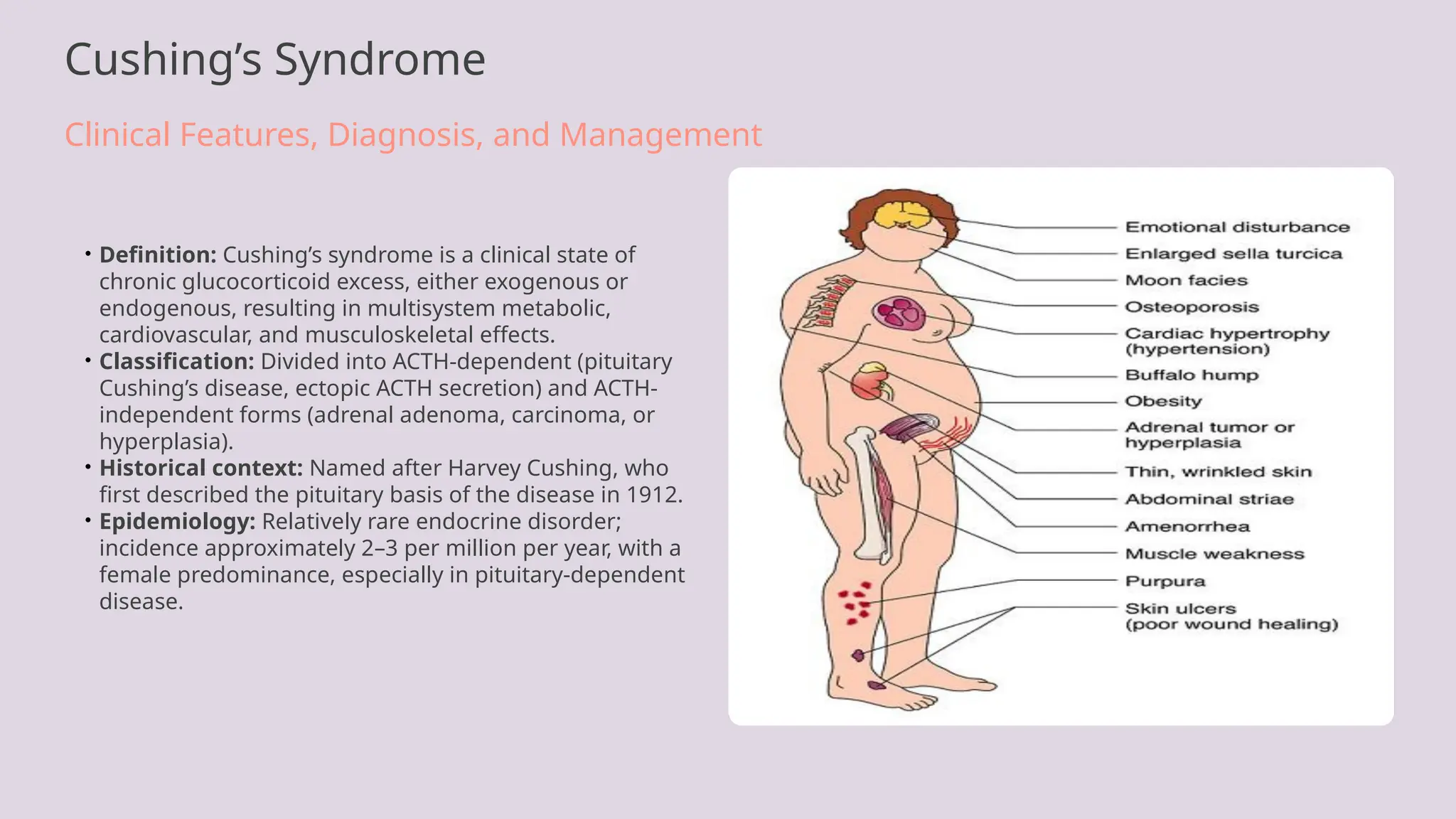
• Definition: Cushing’s syndrome is a clinical state of
chronic glucocorticoid excess, either exogenous or
endogenous, resulting in multisystem metabolic,
cardiovascular, and musculoskeletal effects.
• Classification: Divided into ACTH-dependent (pituitary
Cushing’s disease, ectopic ACTH secretion) and ACTH-
independent forms (adrenal adenoma, carcinoma, or
hyperplasia).
• Historical context: Named after Harvey Cushing, who
first described the pituitary basis of the disease in 1912.
• Epidemiology: Relatively rare endocrine disorder;
incidence approximately 2–3 per million per year, with a
female predominance, especially in pituitary-dependent
disease.
2.
Definition and Classificationof Cushing’s Syndrome
Understanding Endogenous and Exogenous Causes
•Definition: Cushing’s syndrome refers to the clinical consequences of chronic
exposure to excess glucocorticoids, either endogenous (from cortisol overproduction)
or exogenous (from therapeutic corticosteroid use).
•Endogenous Cushing’s Syndrome: Occurs due to overproduction of cortisol by the
adrenal glands. Subdivided into ACTH-dependent and ACTH-independent causes.
•ACTH-Dependent: Includes pituitary adenomas (Cushing’s disease) responsible for
~70% of cases, and ectopic ACTH secretion from non-pituitary tumors such as
bronchial carcinoids or small-cell lung carcinoma.
•ACTH-Independent: Arises from adrenal adenomas, carcinomas, or bilateral adrenal
hyperplasia; results in autonomous cortisol secretion with suppressed ACTH levels.
3.
Pathophysiology of Cushing’sSyndrome
Dysregulation of the Hypothalamic–Pituitary–Adrenal (HPA) Axis
•Normal HPA Axis: The hypothalamus secretes corticotropin-releasing hormone (CRH),
stimulating pituitary ACTH release, which in turn drives adrenal cortisol production.
Cortisol exerts negative feedback on both CRH and ACTH.
•Mechanism in Cushing’s Disease: A pituitary corticotroph adenoma autonomously
secretes ACTH, leading to excessive cortisol synthesis despite loss of feedback inhibition.
This results in bilateral adrenal hyperplasia.
•Adrenal Tumor Mechanism: In ACTH-independent forms, a cortisol-secreting adrenal
adenoma or carcinoma bypasses pituitary control, suppressing CRH and ACTH secretion.
•Ectopic ACTH Production: Non-pituitary tumors (e.g., small-cell lung carcinoma, bronchial
carcinoid) produce ACTH or CRH, stimulating the adrenal cortex and causing
hypercortisolism.
4.
Clinical Features ofCushing’s Syndrome
Systemic Manifestations of Chronic Cortisol Excess
•Body Fat Redistribution: Central (truncal) obesity with thin extremities, moon facies,
and dorsocervical fat pad (buffalo hump) due to altered lipid metabolism and insulin
resistance.
•Skin and Connective Tissue: Skin thinning, easy bruising, purple striae (especially on
abdomen and thighs), and delayed wound healing from collagen degradation.
•Musculoskeletal System: Proximal myopathy and osteoporosis from protein
catabolism and calcium loss; increased risk of vertebral fractures.
•Cardiovascular and Metabolic Effects: Hypertension, glucose intolerance,
dyslipidemia, and truncal obesity contribute to metabolic syndrome and increased
cardiovascular risk.
5.
Differentiating True Cushing’sSyndrome from Pseudo-Cushing’s States
Clinical and Biochemical Distinctions
•Pseudo-Cushing’s States: Conditions such as chronic alcoholism, major depressive
disorder, and severe obesity may produce hypercortisolism-like biochemical findings
without autonomous cortisol secretion.
•Pathophysiologic Difference: Pseudo-Cushing’s arises from physiological activation
of the HPA axis, whereas true Cushing’s involves loss of negative feedback control
due to ACTH or adrenal autonomy.
•Clinical Differentiation: Features such as proximal myopathy, wide purple striae,
easy bruising, and hypokalemia are more typical of true Cushing’s syndrome.
•Dynamic Testing: Dexamethasone-CRH test or serial late-night salivary cortisol
measurement helps distinguish pseudo-Cushing’s from mild true Cushing’s disease.
6.
Diagnostic Strategy inSuspected Cushing’s Syndrome
From Clinical Suspicion to Biochemical Confirmation
•Initial Clinical Suspicion: Consider Cushing’s syndrome in patients with progressive
obesity, muscle weakness, hypertension, diabetes, and suggestive stigmata such as
striae and facial plethora.
•Exclude Exogenous Steroid Use: A crucial first step, as iatrogenic corticosteroid
exposure is the most common cause of hypercortisolism.
•Screening Objectives: Establish biochemical evidence of hypercortisolism using at
least two independent screening tests.
•Sequential Approach: Proceed from clinical assessment to initial screening (ONDST,
UFC, late-night salivary cortisol), followed by confirmatory and localization studies.
7.
Screening Tests forCushing’s Syndrome
Establishing Biochemical Evidence of Hypercortisolism
•Overnight Dexamethasone Suppression Test (ONDST): 1 mg dexamethasone given
at 11 pm; serum cortisol measured at 8 am next morning. Cortisol >50 nmol/L (1.8
μg/dL) suggests loss of normal feedback suppression.
•Low-Dose Dexamethasone Suppression Test (LDDST): 2 mg/day for 48 hours;
failure to suppress plasma cortisol indicates Cushing’s syndrome.
•24-Hour Urinary Free Cortisol (UFC): Measures unbound cortisol excretion over 24
hours; values >3× upper limit of normal are diagnostic, provided adequate urine
collection.
•Late-Night Salivary Cortisol: Reflects loss of diurnal variation. Elevated midnight
cortisol (>4.3 nmol/L) supports hypercortisolism diagnosis.
8.
Interpretation of ScreeningResults and Diagnostic Algorithm
Structured Evaluation of Hypercortisolism (Equivalent to Fig. 20.22)
•Confirming Hypercortisolism: If two different screening tests are positive, proceed
to determine the cause. If discordant, repeat testing or exclude pseudo-Cushing’s.
•Measure Plasma ACTH: Low or undetectable ACTH indicates ACTH-independent
Cushing’s (adrenal cause). Normal or elevated ACTH suggests ACTH-dependent
etiology.
•High-Dose Dexamethasone Suppression Test (HDDST): Suppression >50% of
baseline cortisol implies pituitary Cushing’s disease; absent suppression suggests
ectopic or adrenal source.
•CRH Stimulation Test: ACTH and cortisol rise after CRH in pituitary disease but
remain flat in ectopic ACTH or adrenal tumors.
9.
Determining the Causeof Cushing’s Syndrome
ACTH Measurement and Etiologic Classification (Equivalent to Fig. 20.23)
•Plasma ACTH Measurement: First-line test to distinguish ACTH-dependent from
ACTH-independent causes. Low ACTH (<5 pg/mL) indicates adrenal source;
high/normal ACTH (>20 pg/mL) suggests pituitary or ectopic origin.
•ACTH-Independent Cushing’s Syndrome: Caused by adrenal adenoma, carcinoma,
or macronodular hyperplasia; confirmed by adrenal imaging and suppressed ACTH.
•ACTH-Dependent Cushing’s Syndrome: Requires differentiation between pituitary
Cushing’s disease and ectopic ACTH secretion via dynamic and imaging studies.
•Intermediate ACTH Levels: May occur in cyclic Cushing’s or assay interference;
repeat measurements during active disease are essential.
10.
Dynamic Tests forDifferential Diagnosis
High-Dose Dexamethasone, CRH Stimulation, and Inferior Petrosal Sinus Sampling
•High-Dose Dexamethasone Suppression Test (HDDST): Pituitary Cushing’s disease
typically shows >50% suppression of cortisol; ectopic ACTH secretion and adrenal
tumors do not suppress.
•CRH Stimulation Test: Pituitary adenomas respond with a rise in ACTH and cortisol
after CRH, while ectopic ACTH sources show minimal or absent response.
•Inferior Petrosal Sinus Sampling (IPSS): Gold standard for differentiating pituitary
from ectopic ACTH production. Central-to-peripheral ACTH ratio >2 at baseline or >3
post-CRH indicates pituitary origin.
•Interpretation Caveats: Technical precision and experienced centers are crucial.
False negatives may occur due to venous asymmetry or cyclic disease.
11.
Imaging Modalities inCushing’s Syndrome
Radiologic Localization After Biochemical Diagnosis
•Pituitary MRI: High-resolution gadolinium-enhanced MRI detects pituitary
microadenomas in 60–70% of Cushing’s disease cases. Lesions <2 mm may be
missed, requiring correlation with IPSS.
•Adrenal Imaging: CT or MRI of adrenal glands identifies adenomas, carcinomas, or
hyperplasia in ACTH-independent Cushing’s syndrome. Adenomas appear as small,
homogeneous, lipid-rich lesions.
•Ectopic ACTH Source Localization: Chest and abdominal CT, octreotide or PET scans
help locate ectopic ACTH-producing tumors such as bronchial carcinoids or small-cell
lung cancer.
•Imaging Sequence: Imaging should only follow biochemical confirmation to avoid
incidentaloma-related misinterpretation.
12.
General Principles ofManagement in Cushing’s Syndrome
Multidisciplinary and Etiology-Specific Approach
• Treatment Objectives: Normalize cortisol levels, reverse
clinical features, treat underlying tumor, and manage
comorbidities such as diabetes, hypertension, and
osteoporosis.
• Preoperative Preparation: Control severe
hypercortisolism with medical therapy (e.g., metyrapone,
ketoconazole, osilodrostat) and optimize metabolic and
cardiovascular function.
• Definitive Management: Pituitary surgery for Cushing’s
disease, adrenalectomy for adrenal tumors, and
resection of ectopic ACTH-secreting tumors when
feasible.
• Multidisciplinary Care: Endocrinologists,
neurosurgeons, anesthesiologists, and intensivists
coordinate perioperative care and long-term follow-up.
13.
Cushing’s Disease (PituitaryOrigin)
Pathogenesis and Surgical Management
• Pathogenesis: Caused by a pituitary corticotroph adenoma secreting ACTH autonomously, resulting in
bilateral adrenal hyperplasia and cortisol overproduction.
• Clinical Profile: More common in women (5:1). Features include progressive weight gain, amenorrhea,
and signs of cortisol excess.
• Definitive Treatment: Transsphenoidal adenomectomy is the treatment of choice; performed under MRI
guidance by experienced neurosurgeons.
• Remission and Recurrence: Initial remission rates 70–90%; long-term recurrence occurs in up to 25% and
necessitates lifelong follow-up.
14.
Adrenal Tumours inCushing’s Syndrome
Adrenal Adenoma, Carcinoma, and Hyperplasia
• Adrenal Adenoma: Benign, unilateral, cortisol-secreting
tumors accounting for most ACTH-independent
Cushing’s cases. Typically <4 cm with low attenuation on
CT.
• Adrenal Carcinoma: Rare but aggressive; presents with
severe hypercortisolism, virilization, and large
heterogeneous adrenal mass. Prognosis poor despite
surgery.
• Primary Bilateral Macronodular Hyperplasia:
Characterized by multiple nodules in both adrenal
glands; may involve aberrant hormone receptor
expression.
• Surgical Treatment: Laparoscopic adrenalectomy is
curative for adenoma; open resection required for
carcinoma with careful perioperative glucocorticoid
coverage.
15.
Ectopic ACTH Syndrome
ParaneoplasticSources of ACTH and CRH
• Definition: Ectopic ACTH syndrome arises from non-
pituitary tumors producing ACTH or CRH, leading to
cortisol excess independent of pituitary control.
• Common Sources: Small-cell lung carcinoma, bronchial
and thymic carcinoid tumors, pancreatic neuroendocrine
tumors, and medullary thyroid carcinoma.
• Clinical Features: Rapid onset of severe
hypercortisolism with profound hypokalemic alkalosis,
hypertension, and muscle wasting; pigmentation may
occur due to elevated ACTH.
• Diagnosis: Distinguished from pituitary disease by lack
of suppression with HDDST or CRH test, high ACTH
levels, and localization via chest/abdominal imaging or
PET.
16.
Summary and ClinicalPearls
Key Insights in Diagnosis and Management of Cushing’s Syndrome
•Early Recognition: Prompt identification of progressive symptoms such as proximal
myopathy, facial rounding, and purple striae enables earlier investigation and
improved outcomes.
•Structured Diagnostic Approach: Follow a sequential algorithm: confirm cortisol
excess determine ACTH dependence localize the source initiate targeted
→ → →
therapy.
•Avoid Diagnostic Pitfalls: Exclude exogenous steroid use and pseudo-Cushing’s
states before biochemical testing to prevent misclassification.
•Individualized Management: Therapeutic strategy depends on etiology, patient
comorbidities, and surgical feasibility; interdisciplinary coordination is essential.
#1 Welcome to this presentation on Cushing’s syndrome, a classic yet challenging endocrine disorder characterized by chronic cortisol excess. This session will review clinical presentation, diagnostic algorithms, and management principles. We begin by defining Cushing’s syndrome and highlighting its key classifications. The syndrome can arise from endogenous cortisol overproduction or from prolonged exposure to exogenous glucocorticoids. Clinicians must differentiate between ACTH-dependent and ACTH-independent forms to direct therapy. Historically, Harvey Cushing’s discovery of pituitary-driven hypercortisolism in 1912 marked a turning point in neuroendocrinology. Though rare, Cushing’s syndrome has significant morbidity and mortality if untreated, underscoring the importance of timely diagnosis and precise management.
#2 Cushing’s syndrome can result from internal or external sources of glucocorticoid excess. Understanding its classification is crucial for directing investigations and treatment. Endogenous cases originate within the body’s HPA axis, while exogenous causes stem from therapeutic steroid use. Among endogenous forms, ACTH-dependent causes such as pituitary adenomas and ectopic ACTH secretion dominate, contrasting with ACTH-independent adrenal sources. Recognizing these distinctions is key before interpreting biochemical results or proceeding with imaging.
#3 The core of Cushing’s syndrome lies in dysregulation of the hypothalamic–pituitary–adrenal axis. Under physiological conditions, cortisol secretion follows a diurnal rhythm and is tightly regulated through negative feedback. In Cushing’s disease, a pituitary adenoma overrides this control, leading to sustained ACTH stimulation and adrenal hyperplasia. In contrast, ACTH-independent forms stem from autonomous adrenal cortisol secretion that suppresses pituitary and hypothalamic input. Rarely, ectopic ACTH or CRH secretion drives the syndrome, often signaling an underlying malignancy. These disruptions culminate in systemic metabolic, cardiovascular, and immunologic consequences typical of hypercortisolism.
#4 Cushing’s syndrome produces a striking constellation of physical signs rooted in cortisol’s catabolic and metabolic actions. Patients typically present with centripetal fat distribution—moon face, truncal obesity, and the buffalo hump—contrasting with wasted limbs. Cortisol-induced skin thinning leads to wide, purple striae and easy bruising. Muscle weakness and osteoporosis are hallmarks of chronic protein and calcium catabolism. In the metabolic domain, cortisol raises blood pressure, glucose, and lipids, driving cardiovascular risk. Neuropsychiatric disturbances and menstrual changes further illustrate the pervasive impact of hypercortisolism across multiple systems.
#5 In clinical practice, several physiological or psychiatric states mimic Cushing’s syndrome. Chronic alcoholism, major depression, and morbid obesity can cause transient activation of the HPA axis leading to elevated cortisol without true autonomy. Differentiating these states is critical to prevent unnecessary invasive testing or surgery. The presence of specific stigmata like muscle wasting, violaceous striae, and spontaneous bruising suggest genuine Cushing’s. Dynamic suppression tests, such as the dexamethasone-CRH test, provide biochemical clarity, and resolution of cortisol excess after treating the primary disorder confirms pseudo-Cushing’s.
#6 The diagnostic journey in Cushing’s syndrome begins with careful clinical evaluation. Because many features are nonspecific, the threshold for testing should be guided by severity and progression of signs. Exogenous corticosteroid exposure must be excluded before considering endogenous causes. Once suspicion is justified, biochemical testing follows a structured protocol. The process involves initial screening to confirm cortisol excess, followed by dynamic testing and imaging for localization. This systematic approach, coupled with interdisciplinary communication, reduces misdiagnosis and guides appropriate intervention.
#7 Screening for Cushing’s syndrome requires sensitive and specific tests that assess cortisol suppression and circadian rhythm. The overnight dexamethasone suppression test (ONDST) and the 24-hour urinary free cortisol measurement are most widely used. The late-night salivary cortisol test offers a noninvasive and highly discriminating alternative. Interpreting results requires awareness of factors that can elevate cortisol transiently, such as depression or alcoholism. Confirming hypercortisolism on at least two separate tests is essential before moving to localization studies.
#8 Once hypercortisolism is confirmed, interpretation of biochemical data follows an algorithmic pathway. The key discriminator is plasma ACTH concentration, which separates ACTH-dependent from independent causes. High-dose dexamethasone suppression and CRH stimulation tests further refine diagnosis by distinguishing pituitary from ectopic ACTH secretion. A stepwise, structured diagnostic algorithm prevents premature imaging and guides targeted evaluation. This logical framework, akin to the algorithm in Fig. 20.22, ensures accurate etiological identification prior to intervention.
#9 Determining the cause of Cushing’s syndrome hinges on plasma ACTH measurement. Suppressed ACTH levels indicate an adrenal source, whereas elevated or normal ACTH suggests ACTH-dependent disease. The next diagnostic steps depend on this division. For ACTH-independent forms, adrenal imaging identifies adenomas or carcinomas. ACTH-dependent hypercortisolism requires dynamic testing to distinguish between pituitary and ectopic sources. Intermediate ACTH values may require repeat testing or sampling during disease activity. This structured approach parallels the diagnostic flow outlined in Fig. 20.23.
#10 Dynamic endocrine testing refines the differentiation between pituitary and ectopic ACTH-dependent Cushing’s syndrome. The high-dose dexamethasone suppression test leverages partial feedback retention in pituitary disease, whereas ectopic sources fail to suppress cortisol. CRH stimulation further confirms pituitary responsiveness. Inferior petrosal sinus sampling, though invasive, remains the diagnostic gold standard, providing definitive biochemical localization when imaging is inconclusive. Accurate interpretation depends on technical expertise and integration of hormonal and radiological data.
#11 Imaging studies in Cushing’s syndrome serve to localize the source of cortisol excess after biochemical confirmation. Pituitary MRI remains the mainstay for identifying microadenomas, though small lesions can evade detection. For ACTH-independent cases, adrenal imaging distinguishes benign adenomas from carcinomas. When ectopic ACTH secretion is suspected, thoracic and abdominal CT or functional imaging such as octreotide scintigraphy or PET scans assist localization. Importantly, imaging must never precede biochemical proof of Cushing’s syndrome, as incidental findings can mislead clinicians.
#12 Effective management of Cushing’s syndrome requires an integrated approach addressing both the biochemical and clinical consequences of cortisol excess. The primary goal is normalization of cortisol levels while treating the underlying etiology. Preoperative medical therapy is often necessary in severe hypercortisolism to reduce perioperative risk. Definitive therapy depends on the source: pituitary surgery for Cushing’s disease, adrenalectomy for adrenal tumors, and targeted resection or medical control for ectopic ACTH secretion. Multidisciplinary coordination and long-term hormonal monitoring are vital to prevent recurrence and manage postoperative adrenal insufficiency.
#13 Cushing’s disease, the pituitary-dependent form of hypercortisolism, represents the majority of endogenous cases. It arises from an ACTH-secreting corticotroph microadenoma that stimulates bilateral adrenal hyperplasia. Transsphenoidal pituitary surgery remains the gold-standard treatment, offering the best chance for cure with preservation of pituitary function. Despite high initial remission rates, recurrence can occur years later, underscoring the need for lifelong hormonal monitoring. For non-surgical candidates or persistent disease, radiotherapy and medical therapies serve as adjunctive options.
#14 Adrenal tumors represent ACTH-independent causes of Cushing’s syndrome. Adrenal adenomas are usually benign, small, and unilateral, whereas adrenal carcinomas are rare but clinically severe. Bilateral hyperplasia introduces additional diagnostic complexity. Imaging via CT or MRI identifies these lesions and informs surgical planning. Laparoscopic adrenalectomy offers definitive cure for adenomas, while carcinomas often require open surgery and have a guarded prognosis. Postoperative cortisol monitoring is vital, as transient adrenal insufficiency commonly follows curative resection.
#15 Ectopic ACTH syndrome represents a paraneoplastic variant of Cushing’s syndrome in which non-pituitary tumors secrete ACTH or CRH. The most frequent causes are small-cell lung carcinoma and bronchial carcinoid tumors. Clinical presentation is often acute and severe, with striking hypokalemia and metabolic alkalosis. Diagnostic differentiation from pituitary disease relies on dynamic testing and imaging. Treatment targets the underlying tumor, but interim medical management with cortisol-lowering agents is frequently necessary due to the rapid biochemical impact of ectopic ACTH production.
#16 Cushing’s syndrome exemplifies the complexity of endocrine disorders requiring integrated clinical reasoning. Early recognition and systematic testing can transform outcomes, while failure to exclude exogenous or pseudo-Cushing’s states risks misdiagnosis. The diagnostic algorithm—from biochemical confirmation to localization—must be meticulously followed. Management strategies vary by etiology and demand multidisciplinary expertise. Even after apparent cure, patients require vigilant long-term follow-up to detect recurrence and address persistent comorbidities.