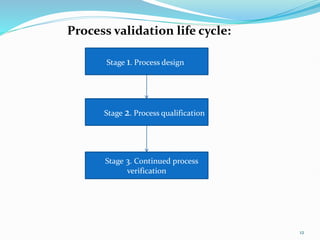
The document discusses validation of pharmaceutical processes. It defines validation as establishing documented evidence that a process will consistently produce a product meeting predetermined specifications. There are several types of validation including process validation to ensure consistency in manufacturing, cleaning validation to minimize contamination, equipment validation to ensure equipment works correctly, and validation of analytical methods to establish test method performance. Proper documentation is essential for validation in the form of validation master plans, protocols, reports and standard operating procedures.