Download as PDF, PPTX














































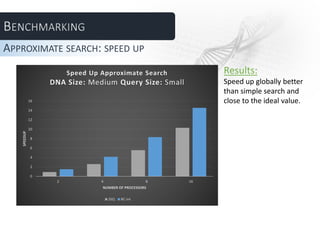
![Benchmarking consisted in evaluating and comparing running
times of each algorithm as function of the following
parameters
• Number of processors (# workers +1) [2, 4, 8, 16]
• DNA length (Small -5MB-, Medium -149 MB-, Large -292MB-)
• Query length (Small -8byte-, Medium -32byte-, Large -64byte-)
• # best allignments -Approximate search only- (10, 50, 100)
In grey the fixed value for the parameter when not evaluated
TEST PLAN](https://image.slidesharecdn.com/presdnadefinitivaaggiustata-130628072327-phpapp02/85/Parallel-DNA-Sequence-Alignment-49-320.jpg)















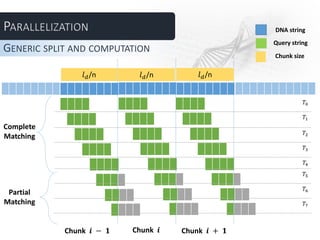
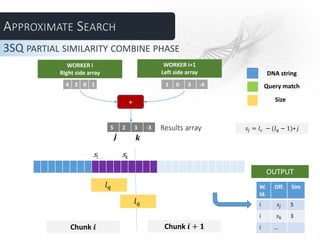
The document describes several parallel approaches for speeding up sequence alignment. It discusses splitting a DNA string into chunks that are distributed to worker nodes. Various techniques are proposed for handling matches that span multiple chunks, including using bigger chunks, on-demand requests for additional data, and having workers find partial matches along chunk edges to be combined by the master. The approaches are analyzed in terms of advantages and disadvantages, and a test plan is outlined to evaluate performance under varying parameters like number of workers and query length.


































































