Osteogenesis imperfecta
Cam Kemikhastalığı (Osteogenesis Imperfecta) nedir?
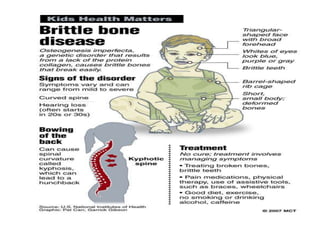
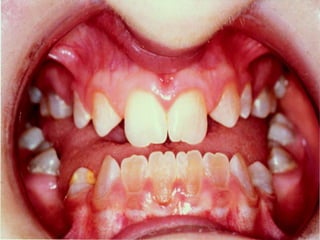
Cam kemik hastalığı kemiklerde kolay ve sık kırılmanın yanı
sıra mavi sklera diş bozuklukları ve işitme bozukluklarının
da birlikte görülebildiği bir hastalıktır.
Cam Kemik hastalığının nedenleri nelerdir?
Cam kemik hastalığı kemik yapısında da bulunan tip 1
kollajenin yapı bozukluğu ile ortaya çıkmaktadır.
2.
• Cam Kemikhastalığında kalıtım faktörü
Cam kemik hastalığı ailevi geçiş gösterdiği gibi anne ve
babanın genetik yapısı normal olduğu halde anne rahminde
iken oluşan mutasyonlar sonucu bebekte hastalığa
sebebiyet verebilir.
Ailevi geçişte anne veya babadan biri hastalığa sahipse
çocukta hastalık riski % 50 dir. Bunun yanı sıra mozaism
denilen tabloda anne ve babanın genlerinin bir kısmında bu
hastalık olmakta anne ve baba sağlam olmaktadır.
Eğer bu hastalıklı genler çocuğa geçerse bu da hastalığa
sebebiyet verebilmektedir. Mozaismi olan anne ve
babalarda hastalıklı çocuğa sahip olma riski % 2-7
arasındadır.
3.
• Cam Kemikhastalığının teşhisi
Cam kemik hastalığını teşhisi klinik olarak
koyulabilmektedir. Ayrıca cilt biyopsisi kanda
genetik test ve intrauterin 14-18 haftalarda
amniosentez ile de teşhis edilebilmektedir.
4.
• Cam Kemikhastalığının çeşitleri
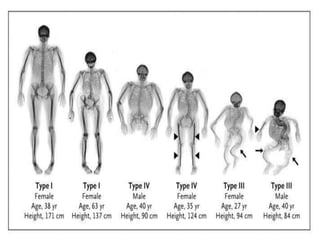
Cam kemik hastalığını başlıca 4 tipi mevcuttur.
Tip 1 Cam Kemik hastalıklı çocuklar doğum
sonrası erken dönemde kaybedilmektedirler.
Yine bu tiplerde Skleraların (göz beyazlarının)
beyaz veya mavi olduğu tiplerin yanında diş
bozukluğu olan yada olmayan işitme
bozukluğu ile seyreden cam kemik hastalığı
çeşitleri mevcuttur.
6.
• Cam Kemikhastalığının gelişimi
Cam kemik hastalığı tip 1 kollajen yapısında
bozukluk şeklinde çocukluk çağında bulgu
vermektedir. Çoğu zaman ilk bulgu tekrarlayan
kırıklar mavi sklera diş bozuklukları şeklinde
görülmektedir. Tip 4 formunda hastalık hafif
seyretmekte ve bazı hastalarda tekrarlayan
kırıklar görülmemektedir.
7.
• Cam Kemikhastalığının tedavisi
Cam kemik hastalığında kırık olunca kırık tedavisi
yapılmalı gecikmiş olgularda gözlenen
deformiteler düzeltilmelidir. Bunun yanısıra gen
tedavisi ve büyüme hormonu ile tedavi
konularında çalışmalar sürmektedir. Son yıllarda
kırık sıklığını azaltmak için osteoporozda da
kullanılan bifosfonatların belli doz ve aralıklarla
cam kemik hastalıklı çocuklara verilmesi ile
başarılı sonuçlar bildirilmektedir. Bu çocukların
mobilizasyonu konusunda fizik tedavi ve
rehabilitasyona mutlak ihtiyaç vardır.
8.
• Dünyadaki hastasayısı
Amerika Birleşik Devletleri'ndeki kesin hasta
sayısı da bilinmemekle birlikte Osteogenesis
İmperfecta Foundation tarafından 20000 ile
50000 arasında cam kemik hastalıklı kişi
olduğu öngörülmektedir.
10.
• Osteogenesis imperfecta(OI) is a group of
genetic disorders that mainly affect the bones.
The term "osteogenesis imperfecta" means
imperfect bone formation. People with this
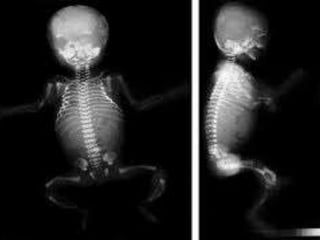
condition have bones that break easily, often
from mild trauma or with no apparent cause.
Multiple fractures are common, and in severe
cases, can occur even before birth. Milder
cases may involve only a few fractures over a
person's lifetime.
11.
• There areat least eight recognized forms of
osteogenesis imperfecta, designated type I
through type VIII. The types can be distinguished
by their signs and symptoms, although their
characteristic features overlap. Type I is the
mildest form of osteogenesis imperfecta and type
II is the most severe; other types of this condition
have signs and symptoms that fall somewhere
between these two extremes. Increasingly,
genetic factors are used to define the different
forms of osteogenesis imperfecta
12.
• The milderforms of osteogenesis imperfecta,
including type I, are characterized by bone
fractures during childhood and adolescence
that often result from minor trauma. Fractures
occur less frequently in adulthood. People
with mild forms of the condition typically have
a blue or grey tint to the part of the eye that is
usually white (the sclera), and may develop
hearing loss in adulthood. Affected individuals
are usually of normal or near normal height.
13.
• How commonis osteogenesis imperfecta?
• This condition affects an estimated 6 to 7 per
100,000 people worldwide. Types I and IV are
the most common forms of osteogenesis
imperfecta, affecting 4 to 5 per 100,000
people.
14.
• What genesare related to osteogenesis imperfecta?
• Mutations in the COL1A1, COL1A2, CRTAP, and LEPRE1
genes cause osteogenesis imperfecta.
• Mutations in the COL1A1 and COL1A2 genes are
responsible for more than 90 percent of all cases of
osteogenesis imperfecta. These genes provide
instructions for making proteins that are used to
assemble type I collagen. This type of collagen is the
most abundant protein in bone, skin, and other
connective tissues that provide structure and strength
to the body.
15.
• Most ofthe mutations that cause osteogenesis
imperfecta type I occur in the COL1A1 gene. These
genetic changes reduce the amount of type I collagen
produced in the body, which causes bones to be brittle
and to fracture easily. The mutations responsible for
most cases of osteogenesis imperfecta types II, III, and
IV occur in either the COL1A1 or COL1A2 gene. These
mutations typically alter the structure of type I collagen
molecules. A defect in the structure of type I collagen
weakens connective tissues, particularly bone,
resulting in the characteristic features of osteogenesis
imperfecta.
16.
• Mutations inthe CRTAP and LEPRE1 genes are responsible for rare,
often severe cases of osteogenesis imperfecta. Cases caused by
CRTAP mutations are usually classified as type VII; when LEPRE1
mutations underlie the condition, it is classified as type VIII. The
proteins produced from these genes work together to process
collagen into its mature form. Mutations in either gene disrupt the
normal folding, assembly, and secretion of collagen molecules.
These defects weaken connective tissues, leading to severe bone
abnormalities and problems with growth.
• In cases of osteogenesis imperfecta without identified mutations in
the COL1A1, COL1A2, CRTAP, or LEPRE1 gene, the cause of the
disorder is unknown. These cases include osteogenesis imperfecta
types V and VI. Researchers are working to identify additional genes
that may be responsible for these conditions.
17.
• How dopeople inherit osteogenesis imperfecta?
• Most cases of osteogenesis imperfecta have an
autosomal dominant pattern of inheritance, which
means one copy of the altered gene in each cell is
sufficient to cause the condition. Many people with
type I or type IV osteogenesis imperfecta inherit a
mutation from a parent who has the disorder. Most
infants with more severe forms of osteogenesis
imperfecta (such as type II and type III) have no history
of the condition in their family. In these infants, the
condition is caused by new (sporadic) mutations in the
COL1A1 or COL1A2 gene.
21.
• Dominance ingenetics is a relationship between
alleles of a single gene, in which one allele masks
the phenotypic expression of another allele at the
same gene locus
• If two alleles of a given gene are identical, the
organism is called a homozygote and is said to be
homozygous with respect to that gene; if instead
the two alleles are different, the organism is a
heterozygote and is heterozygous
22.
A gene mutationis a permanent change in the
DNA sequence that makes up a gene.
Mutations range in size from a single DNA
building block (DNA base) to a large segment
of a chromosome.
23.
• Can wereally believe that we are living a good
life, an ethically decent life if we don't do
anything serious to help reduce poverty
around the world and help save the lives of
children or adults who are likely to die if we
don't increase the amount of aid we are
giving.