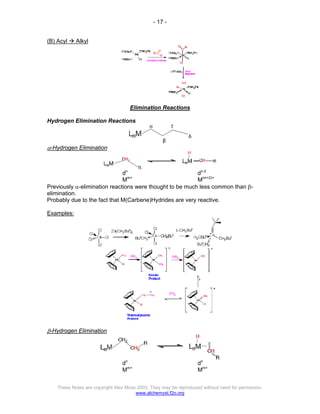
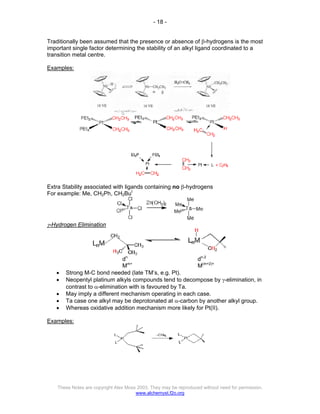
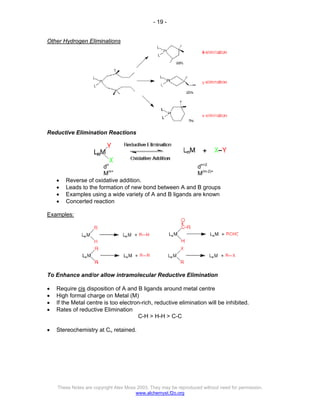
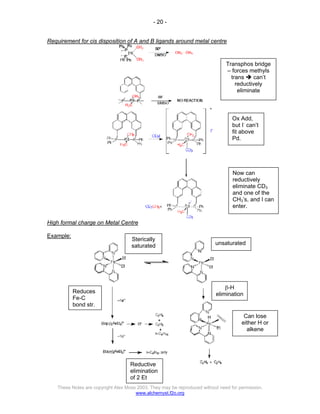
The document discusses the 18 electron rule in organometallic chemistry, explaining how stable organometallic complexes are formed based on the total electron count from metals and ligands. It categorizes complex behaviors into classes regarding electron counts, outlines exceptions, and details factors influencing bonding and reactivity, including mechanisms for ligand insertion and oxidative addition. Several experimental techniques, such as infrared spectroscopy, are mentioned to examine the stereochemistry and kinetics of reactions involving various metal-ligand interactions.








![- 8 -
These Notes are copyright Alex Moss 2003. They may be reproduced without need for permission.
www.alchemyst.f2o.org
The consequence of this is that all bonding interactions are in the equatorial plane.
Mechanisms and Reaction Types
Association and Dissociation:
[M] + L Æ [M]-L
Typically results in the valence electron count increasing by 2, but Oxidation State and
d-count remain the same.
Substitution:
[M]-L + L’ Æ [M]-L’ + L
Net no change in valence electrons, d-count or Oxidation State. Typically proceeds via
first Dissociation, since a 20e intermediate would be highly unstable (occupation of σ*
orbitals required).
Oxidation and Reduction:
Gain or loss of electrons. For oxidation, leads to change in valence electron count (-1),
oxidation state (+1) and d-count (-1).
Migratory Insertion
[M]-A + B Æ [M]-B-A
No net change in electron counts or oxidation states. Classic Example: Me insertion into
Mn carbonyl compound. Proceeds via migration of ligand, then insertion of B.
Formal Definition:
Insertion of an unsaturated ligand (Y) into an adjacent M-X bond.
Mn+
dn
[dn-2
] Mn+
dn
Y = CO, C2H4, C2H2, Benzene, Cyclopentadienyl
X = H, R, Ar, C(O)R
L = Lewis Base, Solvent, PR3, NR3, R2O](https://image.slidesharecdn.com/organometallics-241119184731-0ca5f881/85/Organometallics-Lecture-Notes-by-Chemistry-8-320.jpg)


![- 11 -
These Notes are copyright Alex Moss 2003. They may be reproduced without need for permission.
www.alchemyst.f2o.org
Result 100% cis product
Does not prove either carbonyl insertion or methyl migration.
Experiment 3
Result: Trans
1
Cis
2
Proved using intensities of 1976 cm-1
(cis) and 1949 cm-1
(trans) in I.R. spectrum
• Only migration can give correct product distribution.
• Cannot get trans isomer by de-insertion of CO.
Rate Law for Carbonylation Reaction
Under Normal conditions the following rates are observed
k1 is rate determining, i.e. 1st Order in alkyl–Mn
0th
Order in L (incoming ligand)
if k2 is >>> k-2, then,
High [L]: First order in starting material
Low [L]: Mixed order kinetics
Rate constants are found to be identical for the substitution of CO in Me-CO-Mn(CO)5
and for the decarbonylation of the acetyl compound, i.e. they have the same rate
determining step.
Rate accelerated by Lewis Acid catalysis, e.g. H+
, AlCl3, BF3.
Lewis acids are presumed to stabilise Transition state as shown:](https://image.slidesharecdn.com/organometallics-241119184731-0ca5f881/85/Organometallics-Lecture-Notes-by-Chemistry-11-320.jpg)
![- 12 -
These Notes are copyright Alex Moss 2003. They may be reproduced without need for permission.
www.alchemyst.f2o.org
General Observations of Stereospecificity
Metal Centre
Carbon Centre
Oxidative Addition
[M] + A-B Æ [M](A)(B)
Valence electron count increases by 2, as does Oxidation State. d-count reduces by 2.
Requirements for the Metal Complex
• Metal centre must be d2
or greater.
• Metal centre must be coordinatively unsaturated.
• Metal must have appropriate both empty and vacant orbitals.
Transition Metal Example:
Non-transition Metal Examples:
• Me—Br + Mg Æ Me—Mg—Br
• PF3 + F2 Æ PF5
• SnCl2 + Cl2 Æ SnCl4](https://image.slidesharecdn.com/organometallics-241119184731-0ca5f881/85/Organometallics-Lecture-Notes-by-Chemistry-12-320.jpg)
![- 13 -
These Notes are copyright Alex Moss 2003. They may be reproduced without need for permission.
www.alchemyst.f2o.org
Carbon-Hydrogen Bond Activation
Note: Reaction proceeds faster for C6H6 versus CH4 but Ph—H bond stronger then
CH3—H bond
Kinetic Rate Laws for Oxidative Addition Reactions
(1) d8
Square planar Complexes
e.g. Oxidative addition of XY to IrCl(CO)(PPh3)2 (Vaska's complex)
we observe:
Rate = k2[Ir(I)][XY]
• Simple 2nd Order Kinetics
• Characteristic of Coordinatively Unsaturated systems
(2) d8
Trigonal Bipyramidal Complexes
e.g. Oxidative addition of XY to IrH(CO)(PPh3)3
we observe:
• Characteristic of Coordinatively Saturated complexes
• consistent with the following scheme:](https://image.slidesharecdn.com/organometallics-241119184731-0ca5f881/85/Organometallics-Lecture-Notes-by-Chemistry-13-320.jpg)




![- 22 -
These Notes are copyright Alex Moss 2003. They may be reproduced without need for permission.
www.alchemyst.f2o.org
Molecular Dihydrogen Complexes
Experimental Evidence:
Neutron Diffraction
(time-consuming method)
Solution NMR
(modern method)
d(H—H) = 0.84 Å
JHD ca. 30 Hz
JHH ca. 30 x 6.66
compare d(H2) = 0.74 Å compare J(H2) ca. 300 Hz
Other examples
Dihydrogen-Dihydride Exchange:
Nucleophilic Attack on Ligand
[M]-A + B Æ [M]-A-B
An example would be attack on Cp (complex net charged) by MeLi, or attack on an
alkene (complex net charged) by H-
.
Syntheses
Alkyls –
a) Transmetallation. Use Mg / Li reagents.
b) Carbonylate anions, [LnM(CO)x]n-
act as nucleophiles to attack e.g. MeI.
c) Form metal hydrides (then use these to attack alkenes and β-eliminate).
d) Oxidative Addition – using e.g. MeI and 16e compound.
Carbonyls –
a) metal + CO – needs low enthalpy of atomisation. Works only for Ni and Fe.
b) Metal halide/salt + reducing agent + CO, e.g. CrCl3 + Al + 6CO gives Cr(CO)6 +
AlCl3.
Sandwich –
Arenes:
a) Fischer-Hafner. Ring + MCl3 + Al + NO2S2O4. Works for 1st
row TMs only.
b) Metal atom / vapour. M(s) + electron gun, then add rings to M (atoms). Works for
M = Ti, Nb, W, Mo + some lanthanides. Helps if R substituents on ring.
Cyclopentadienes:
Easier to make since Cp(-) has degree of ionic interaction. Use salt-elimination routes
from NaCp / LiCp* + metal halide.](https://image.slidesharecdn.com/organometallics-241119184731-0ca5f881/85/Organometallics-Lecture-Notes-by-Chemistry-22-320.jpg)















































