Download as PDF, PPTX















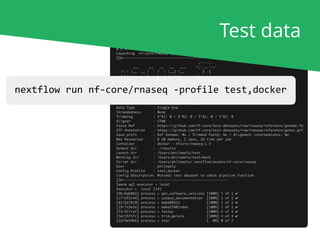
![Running for real
nextflow run nf-core/rnaseq [...]
nf-core launch rnaseq
nf-core download rnaseq
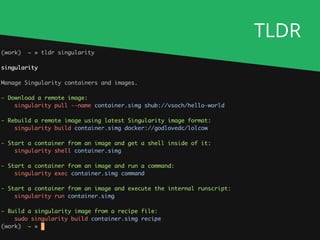
If you've read the documentation..
If not.. (command-line wizard with prompts)
If you're running offline..](https://image.slidesharecdn.com/philewels-nf-coretutorial-190925135352/85/Nextflow-Camp-2019-nf-core-tutorial-20-320.jpg)




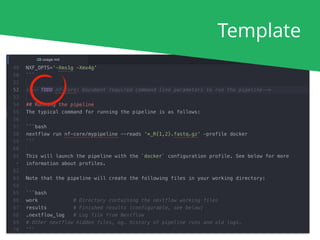
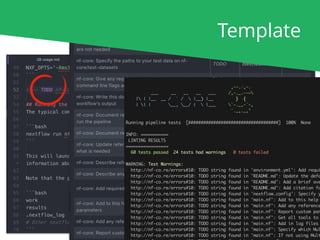

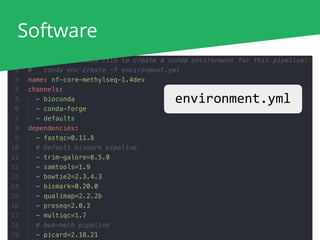


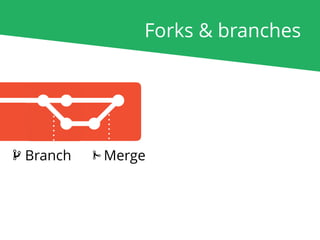
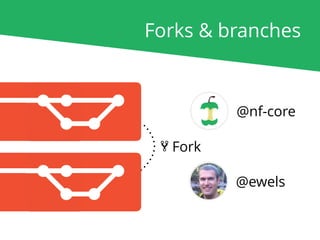
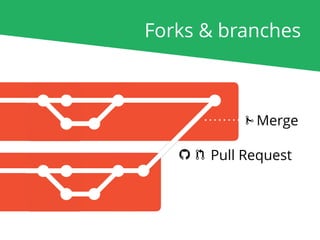







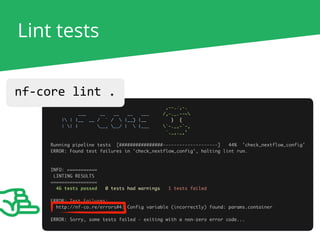
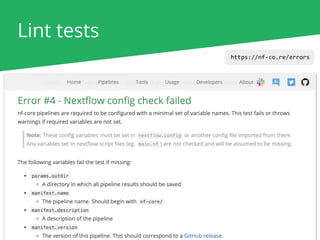










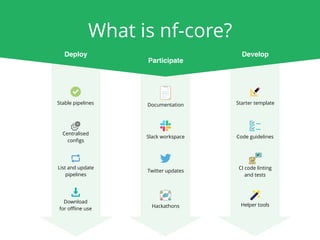
The document outlines the nf-core Nextflow Camp 2019, providing guidance on starting with nf-core tools, including installation, running pipelines, and developing new ones. It presents exercises for users to practice using the nf-core templates, testing pipelines, and releasing their own pipelines. Additionally, it emphasizes community involvement through support platforms and collaboration on GitHub.

















































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)