


![Demographics Estados Unidos Mexicanos - United Mexican States Located in North America between Belize and the US Area: 1,972,550 km 2 Source: CIA - The World Fact book Population: 109,955,400 (7/08 estimate) 31 states + 1 federal district Official language: Spanish Currency: Peso [Conversion: 1 US $ = 12.64 pesos]](https://image.slidesharecdn.com/mptx519finalppttttttt-124841215932-phpapp01/85/My-Presentation-on-Mexico-Medical-Device-Regulations-3-320.jpg)
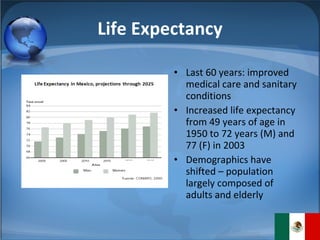


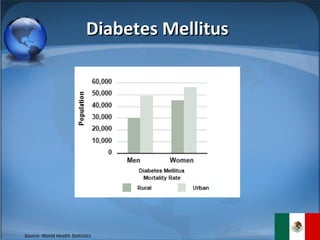
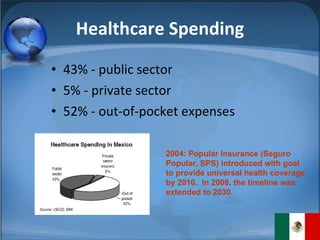














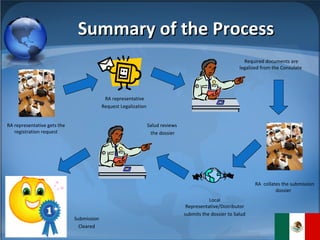




The document summarizes the key considerations for TrojanCare to register their insulin infusion pump in Mexico. It outlines the disease profile, healthcare landscape, market opportunities and regulatory requirements. To register, TrojanCare must submit a dossier to Mexico's health authority including device details, labeling and quality certificates. They also require a local representative for registration and distribution. Overall, Mexico provides opportunities through its large diabetes population and trade agreements, but public coverage favors lower-cost alternatives.




























































![focus_feb10_md_latin-america[1][1]](https://cdn.slidesharecdn.com/ss_thumbnails/2636525a-c68d-4f89-8e4b-36c45678a637-150302131024-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)






