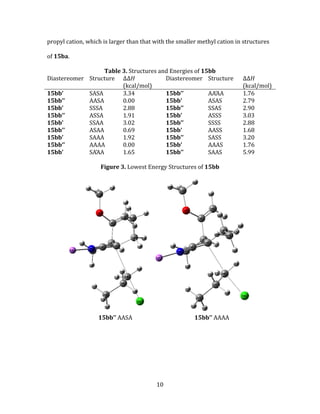
This thesis examines the diastereoselectivity of alkylations of alkoxy-substituted alkenenitriles using density functional theory calculations. Transition state structures were optimized for reactions with various electrophiles. Stereoselectivity is explained by differences in transition state energies, with higher energies corresponding to structures exhibiting torsional strain from gauche orientations of alkyl groups. The position of substituents on the nucleophile and electrophile also influence stereoselectivity by causing steric clashes. Comparing calculated to experimental results allows for better understanding of factors controlling diastereoselectivity in these reactions.