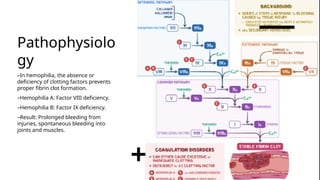
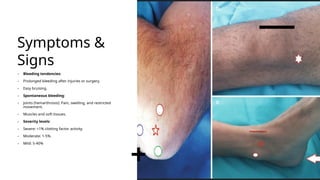
Hemophilia is a rare genetic disorder that affects the blood's ability to clot, primarily manifesting as hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency), mainly affecting males. The condition is inherited in 70% of cases, leading to prolonged bleeding tendencies, and requires careful diagnosis to differentiate from similar bleeding disorders. Treatment options include clotting factor replacement therapy and emerging therapies like gene therapy, which can improve life expectancy and quality of life for affected individuals.