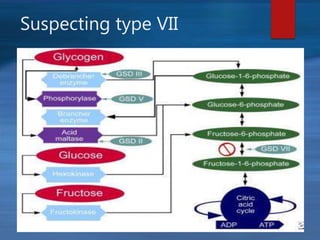
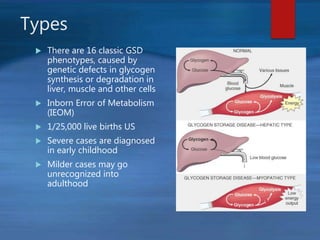
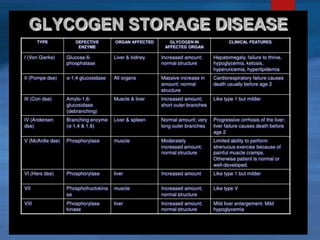
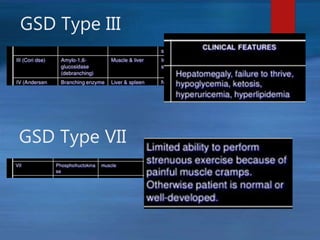
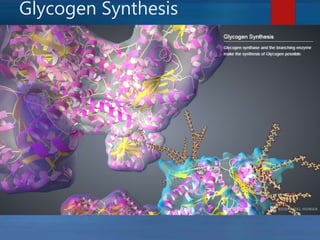
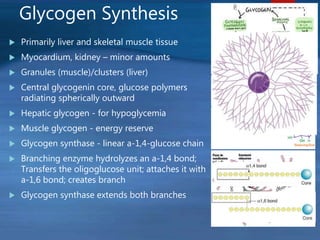
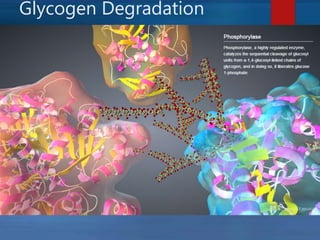
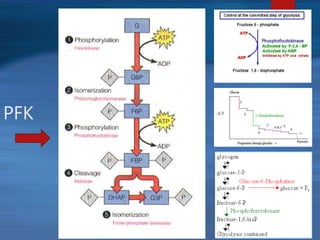
This document describes a case of a novel form of glycogen storage disease (GSD) found in a 62-year-old female patient. Genetic screening revealed a variant in the AGL gene encoding glycogen debranching enzyme, resulting in an amino acid substitution that is believed to impair the enzyme's glucosyltransferase activity specifically in muscle tissue. This causes symptoms like exercise-induced muscle cramps and fatigue consistent with a rare type of GSD IIIc affecting only muscle. The patient responded well to a high-protein diet and carnitine supplementation, representing a newly identified variant of GSD IIId.