Genomic approaches to trypanosome resistance
•
1 like•48 views

Elucidating changes in gene expression in Tryp susceptible and resistant cattle during progression of tryp infection using Affymetrix gene expression Micro arrays
Recommended

More Related Content
What's hot
What's hot (19)
Similar to Genomic approaches to trypanosome resistance
Similar to Genomic approaches to trypanosome resistance (20)
More from Laurence Dawkins-Hall
More from Laurence Dawkins-Hall (20)
Recently uploaded
Recently uploaded (20)
Genomic approaches to trypanosome resistance
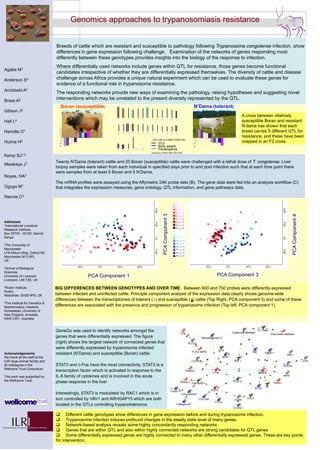
- 1. BIG DIFFERENCES BETWEEN GENOTYPES AND OVER TIME. Between 600 and 750 probes were differently expressed between infected and uninfected cattle. Principle component analysis of the expression data clearly shows genome-wide differences between the transcriptomes of tolerant (×) and susceptible ( ) cattle (Top Right, PCA component 3) and some of these differences are associated with the presence and progression of trypanosome infection (Top left, PCA component 1). This image cannot currently be displayed. This image cannot currently be displayed. Genomics approaches to trypanosomiasis resistance Breeds of cattle which are resistant and susceptible to pathology following Trypanosoma congolense infection, show differences in gene expression following challenge. Examination of the networks of genes responding most differently between these genotypes provides insights into the biology of the response to infection. Where differentially used networks include genes within QTL for resistance, those genes become functional candidates irrespective of whether they are differentially expressed themselves. The diversity of cattle and disease challenge across Africa provides a unique natural experiment which can be used to evaluate these genes for evidence of a functional role in trypanosome resistance. The responding networks provide new ways of examining the pathology, raising hypotheses and suggesting novel interventions which may be unrelated to the present diversity represented by the QTL. GeneGo was used to identify networks amongst the genes that were differentially expressed. The figure (right) shows the largest network of connected genes that were differently expressed by trypanosome infected resistant (N’Dama) and susceptible (Boran) cattle. STAT3 and c-Fos have the most connectivity. STAT3 is a transcription factor which is activated in response to the IL-6 family of cytokines and is involved in the acute phase response in the liver Interestingly, STAT3 is modulated by RAC1 which is in turn controlled by VAV1 and ARHGAP15 which are both located in the QTLs controlling trypanotolerance. Different cattle genotypes show differences in gene expression before and during trypanosome infection. Trypanosome infection induces profound changes in the steady state level of many genes. Network-based analysis reveals some highly concordantly responding networks Genes that are within QTL and also within highly connected networks are strong candidates for QTL genes Some differentially expressed genes are highly connected to many other differentially expressed genes. These are key points for intervention. Twenty N’Dama (tolerant) cattle and 20 Boran (susceptible) cattle were challenged with a lethal dose of T. congolense. Liver biopsy samples were taken from each individual in specified days prior to and post infection such that at each time point there were samples from at least 5 Boran and 5 N’Dama. The mRNA profiles were assayed using the Affymetrix 24K probe sets (B). The gene data were fed into an analysis workflow (C) that integrates the expression measures, gene ontology, QTL information, and gene pathways data. Agaba M1 Anderson S4 Archibald A4 Brass A2 Gibson J5 Hall L4 Hanotte O1 Hulme H2 Kemp SJ1,3 Mwakaya J1 Noyes, HA3 Ogugo M1 Rennie C3 Addresses 1International Livestock Research Institute, Box 30709 - 00100, Nairobi Kenya 2The University of Manchester LF8 Kilburn Bldg, Oxford Rd Manchester M13 9PL UK 3School of Biological Sciences, University of Liverpool, Liverpool, L69 7ZB, UK 4Roslin Institute, Roslin, Midlothian, EH25 9PS, UK 5The Institute for Genetics & Bioinformatics, Hawkins Homestead, University of New England, Armidale, NSW 2351, Australia Acknowledgements: We thank all the staff at the ILRI large animal facility and all colleagues in the Welcome Trust Consortium This work was supported by the Wellcome Trust. QTL with p<0.0043 FDR(10%) PCV Body weight Parasitaemia Hanotte et al PNAS 2003 7443-7448 -15 -10 -5 0 5 10 15 N’Dama (tolerant) -15 -10 -5 0 5 10 15 Boran (susceptible) A cross between relatively susceptible Boran and resistant N’dama has shown that each breed carries 5 different QTL for resistance, and these have been mapped in an F2 cross.