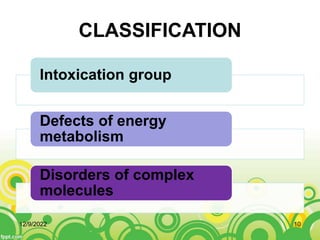
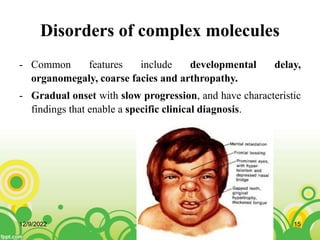
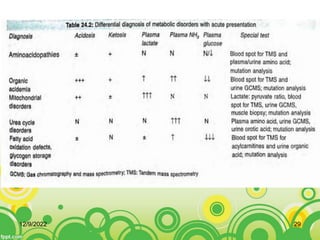
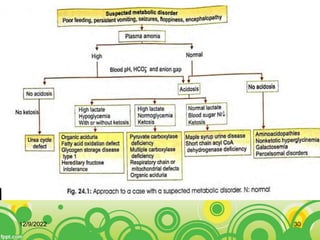
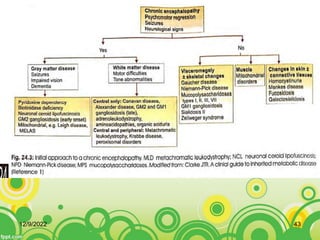
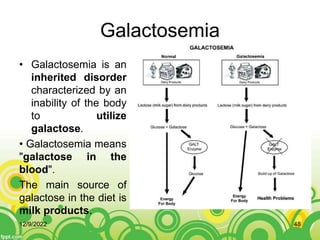
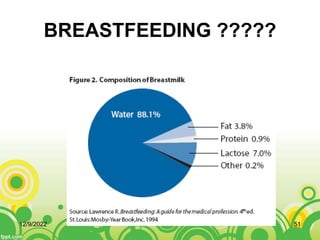
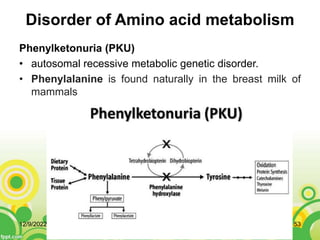
This document provides an overview of inborn errors of metabolism (IEM). It defines IEM as genetic disorders affecting biochemical pathways. The document classifies IEM into 3 main categories: intoxication disorders involving toxic metabolite accumulation, energy metabolism defects, and disorders of complex molecules/organelles. Symptoms of acute and chronic presentation are described. The document outlines diagnostic testing including blood/urine screens and specialized tests. Principles of management are discussed including supportive care. Specific clinical manifestations involving the neurological, hepatic, cardiac and ocular systems are also reviewed.














































![REFERENCES
• Inborn errors of metabolism [Internet]. En.wikipedia.org. 2020 [cited 8 September
2020]. Available from: https://en.wikipedia.org/wiki/Inborn_errors_of_metabolism
• disease M. Maple syrup urine disease: MedlinePlus Medical Encyclopedia
[Internet]. Medlineplus.gov. 2020 [cited 8 September 2020]. Available from:
https://medlineplus.gov/ency/article/000373.htm#:~:text=Maple%20syrup%20urine
%20disease%20(MSUD)%20is%20inherited%2C%20which%20means,these%20c
hemicals%20in%20the%20blood.
• Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s Disease: A Comprehensive Review
of the Molecular Mechanisms. International Journal of Molecular Sciences.
2015;16(12):6419-6431.
• Wilson's disease - Diagnosis and treatment - Mayo Clinic [Internet].
Mayoclinic.org. 2020 [cited 8 September 2020]. Available from:
https://www.mayoclinic.org/diseases-conditions/wilsons-disease/diagnosis-
treatment/drc-20353256
12/9/2022 60](https://image.slidesharecdn.com/geneticsiem-221209053137-c501280f/85/Genetics-IEM-ppt-60-320.jpg)
![REFERENCES
• 5. [Internet]. 2020 [cited 8 September 2020]. Available from:
https://www.researchgate.net/publication/273144908_To_Breastfeed_or_N
ot_PKU_Galactosemia_Other_Rare_Disorders_and_Possible_Misdiagnosi
s [accessed Sep 06 2020]. Available from:
https://www.researchgate.net/publication/273144908_To_Breastfeed_or_N
ot_PKU_Galactosemia_Other_Rare_Disorders_and_Possible_Misdiagnosi
s [accessed Sep 07 2020].
• https://www.researchgate.net/publication/273144908_To_Breastfeed_or_N
ot_PKU_Galactosemia_Other_Rare_Disorders_and_Possible_Misdiagnosi
s [accessed Sep 06 2020].
• Available from:
https://www.researchgate.net/publication/273144908_To_Breastfeed_or_N
ot_PKU_Galactosemia_Other_Rare_Disorders_and_Possible_Misdiagnosi
s [accessed Sep 07 2020].
12/9/2022 61](https://image.slidesharecdn.com/geneticsiem-221209053137-c501280f/85/Genetics-IEM-ppt-61-320.jpg)





































































