Download to read offline


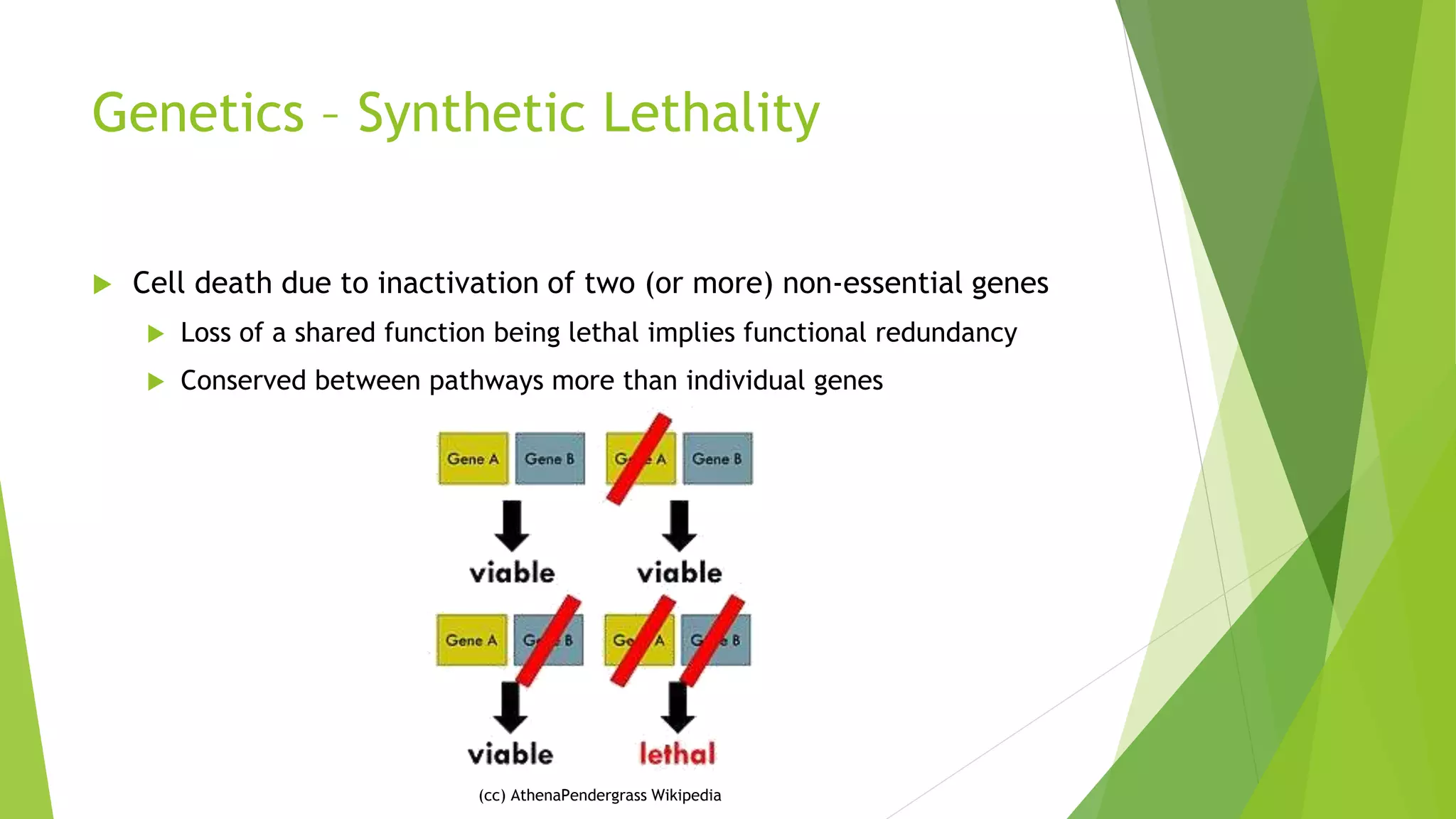
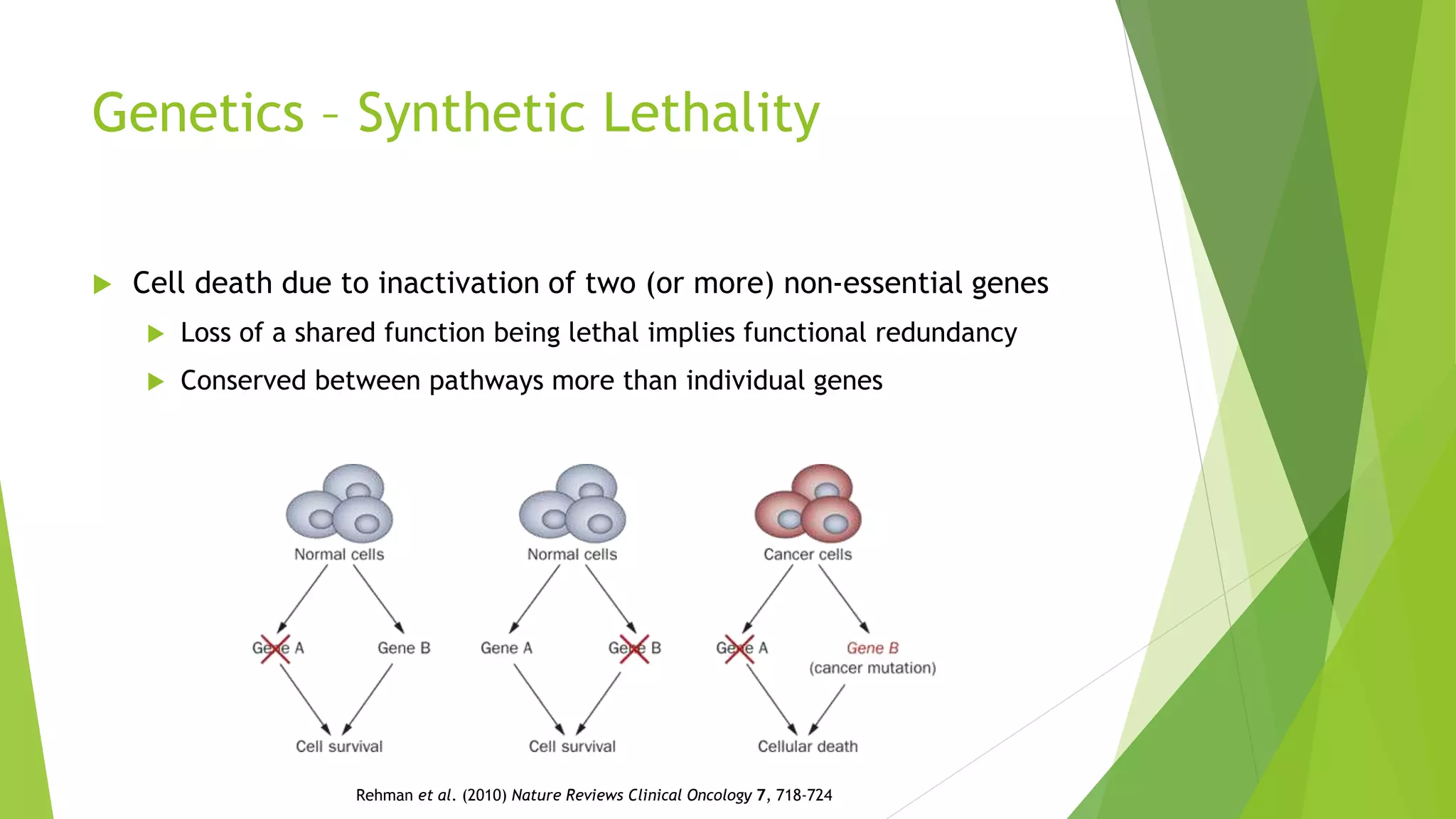




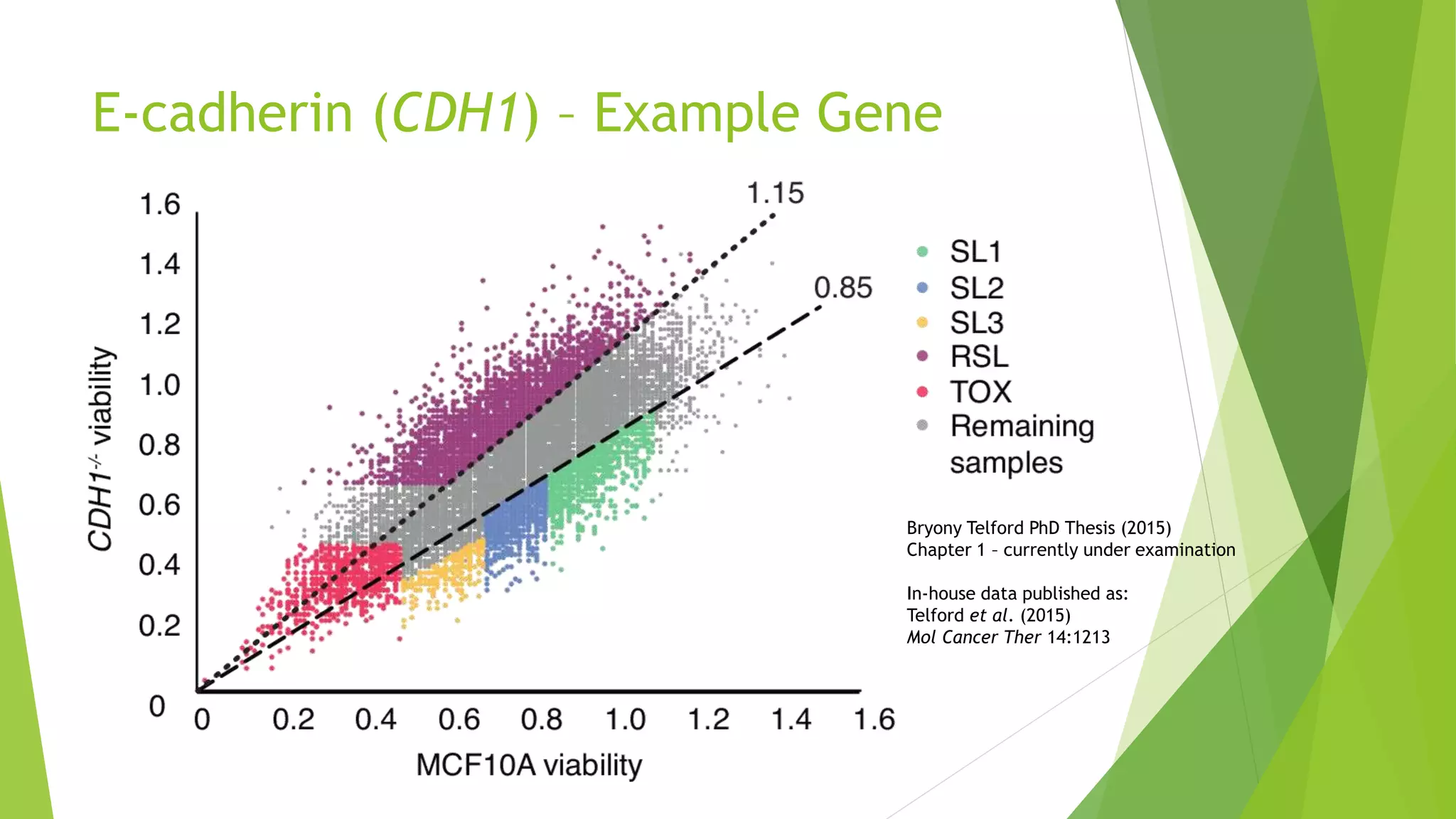

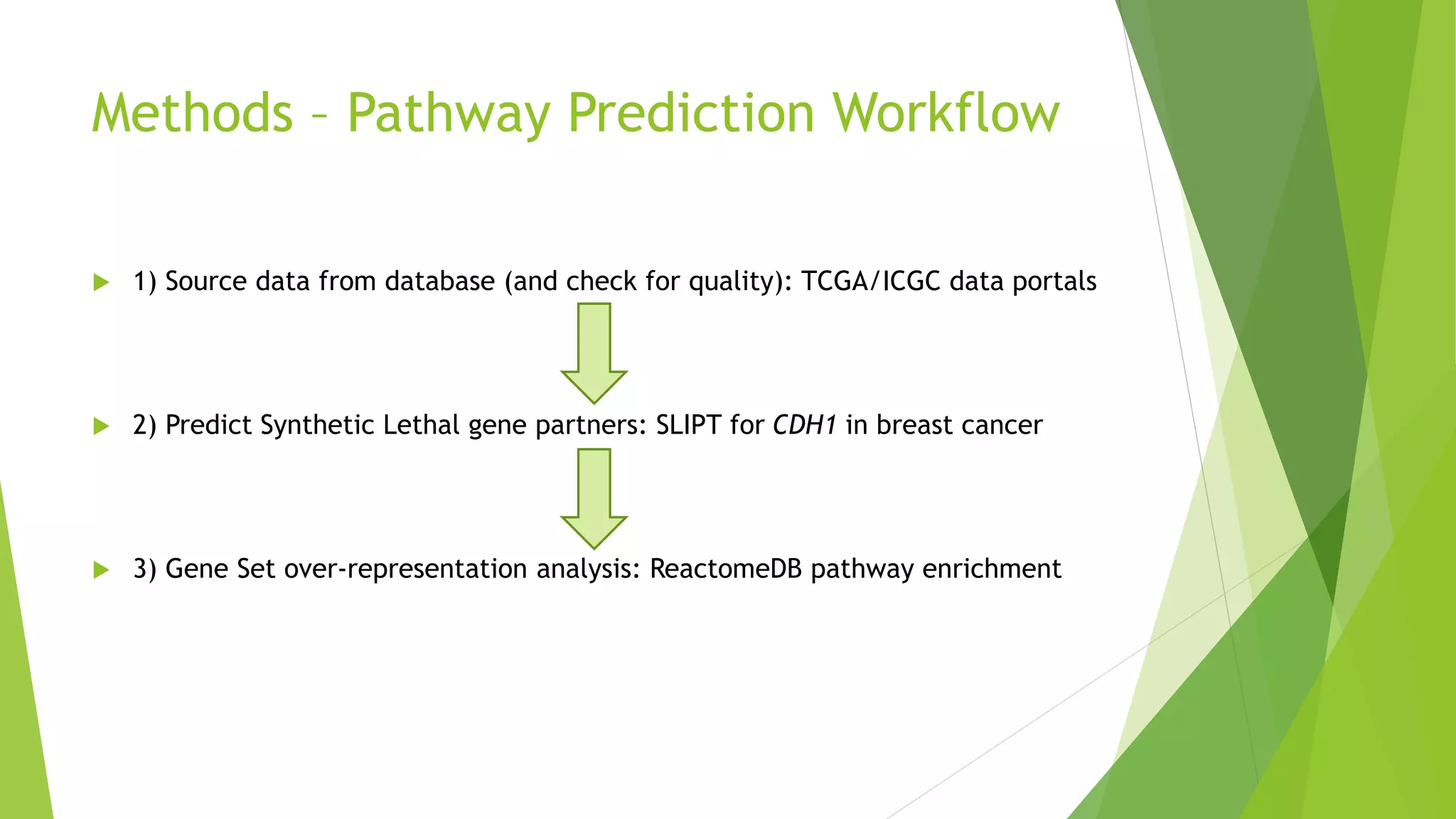
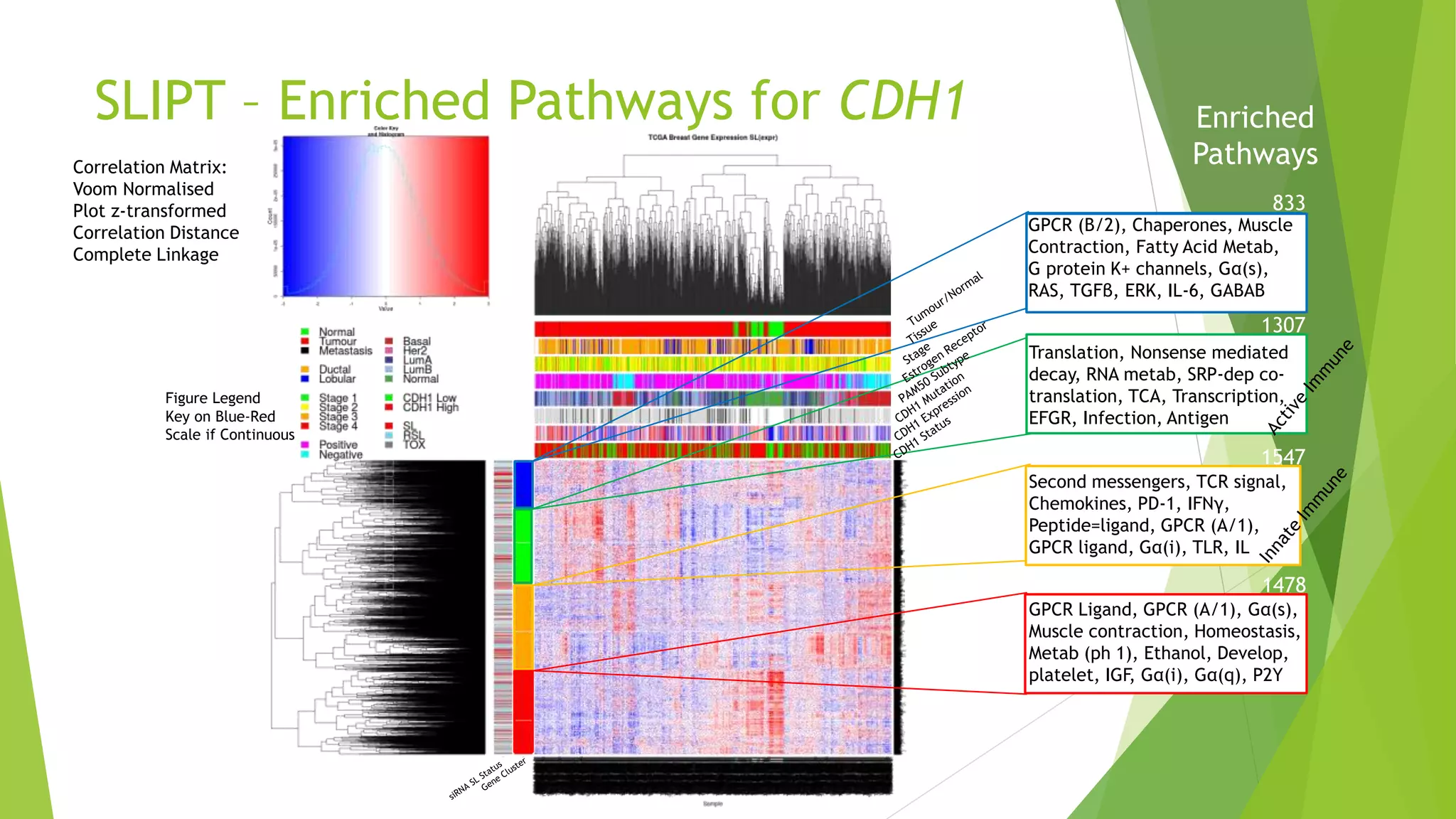
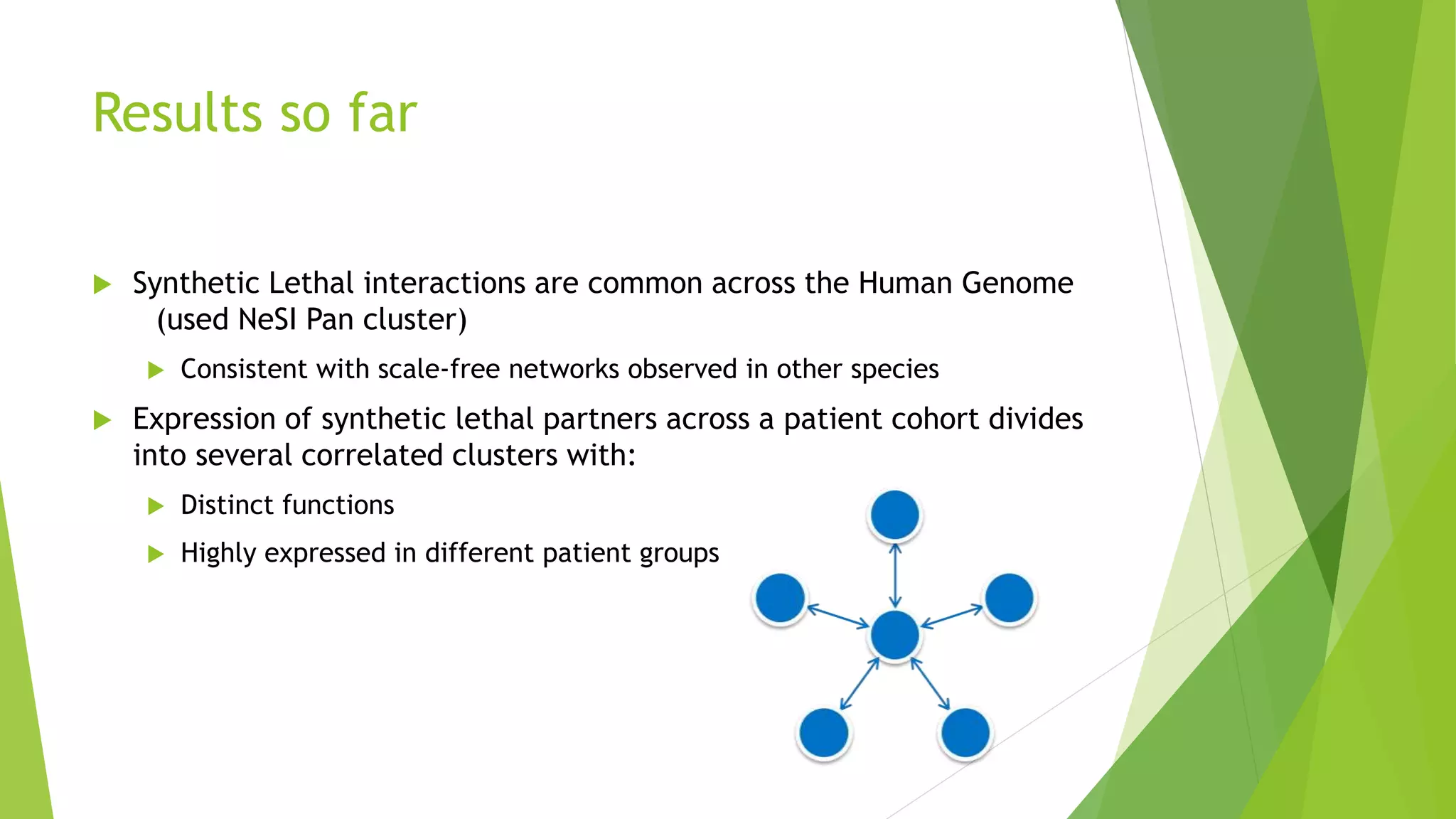
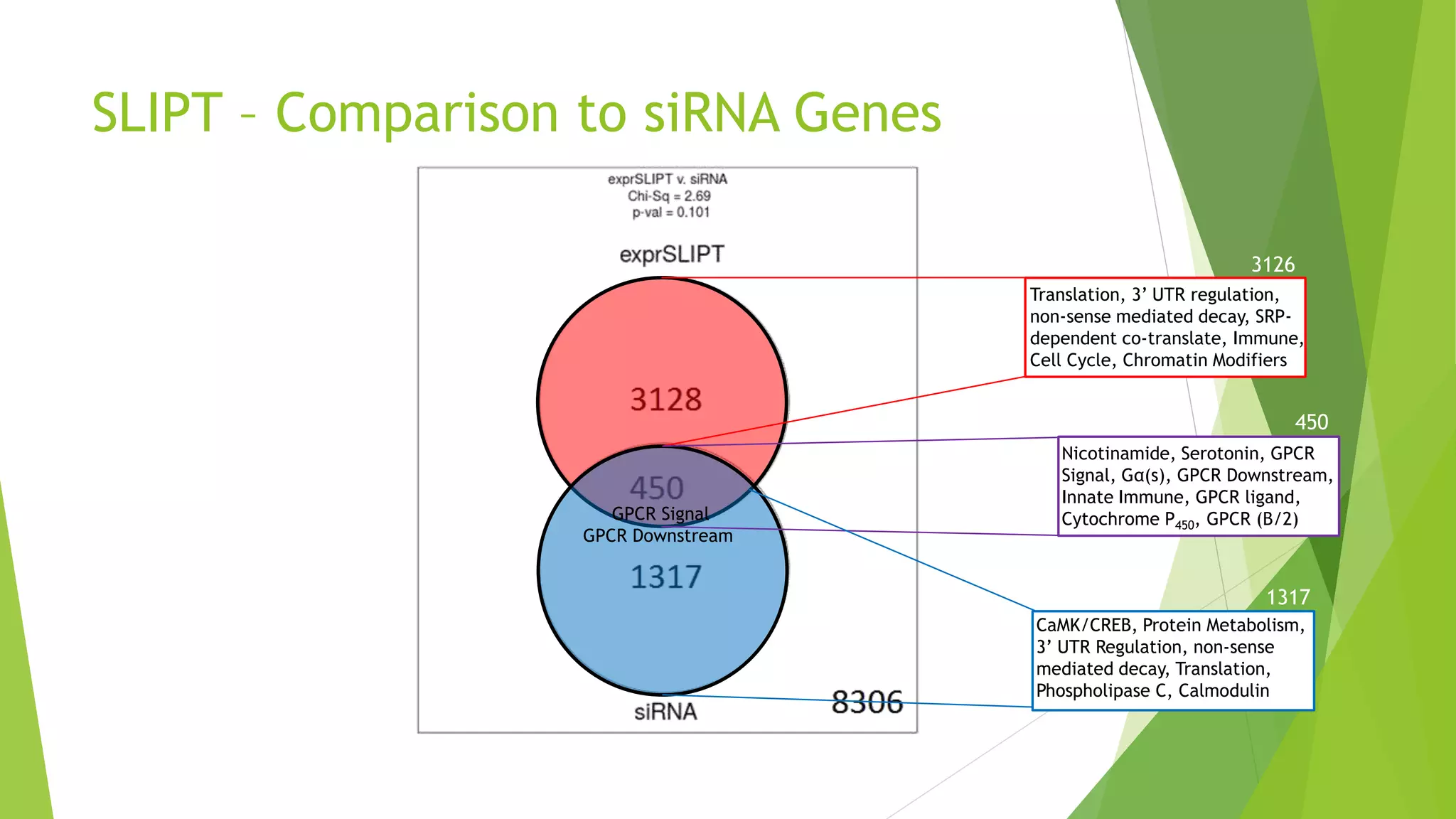



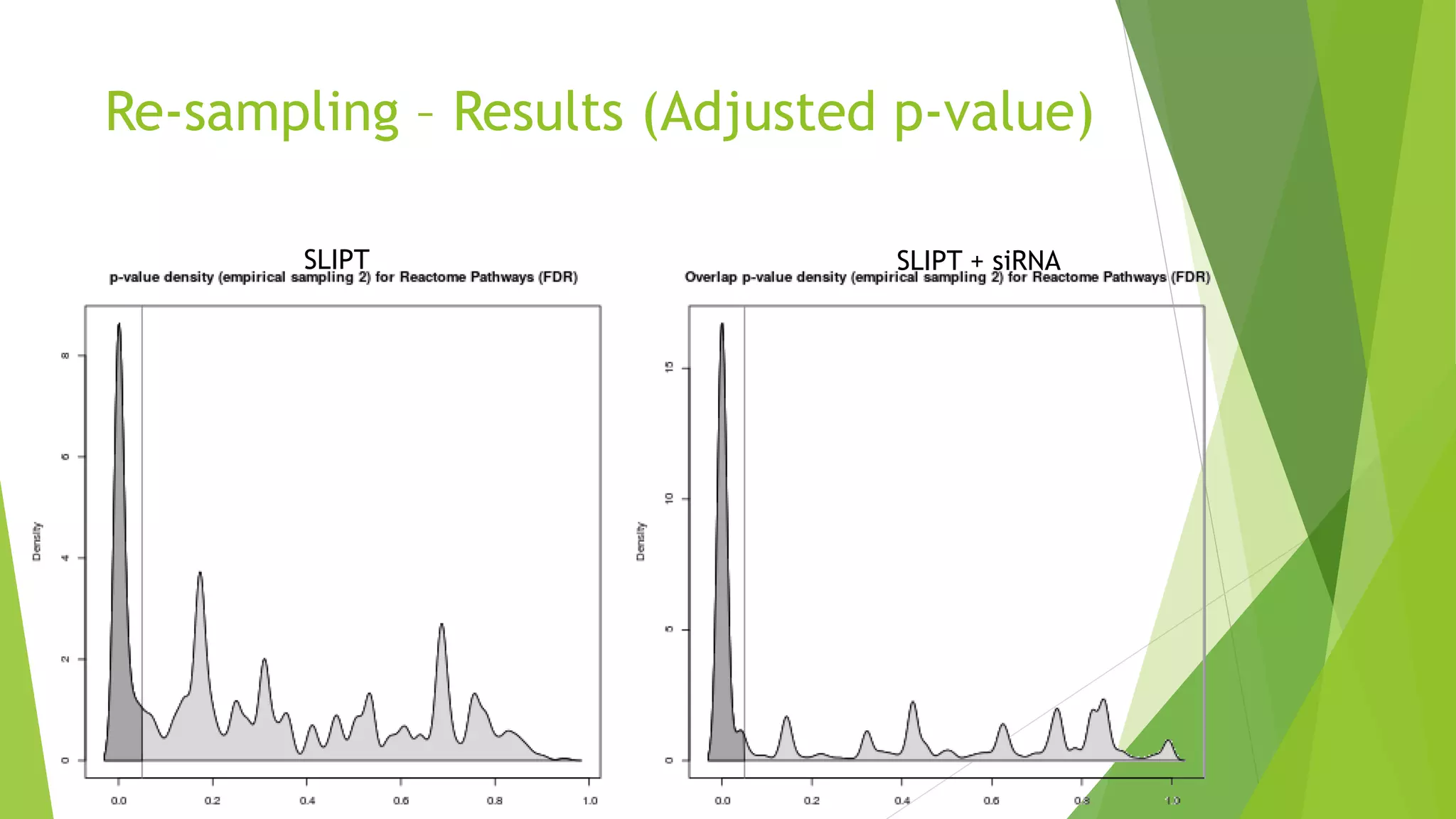

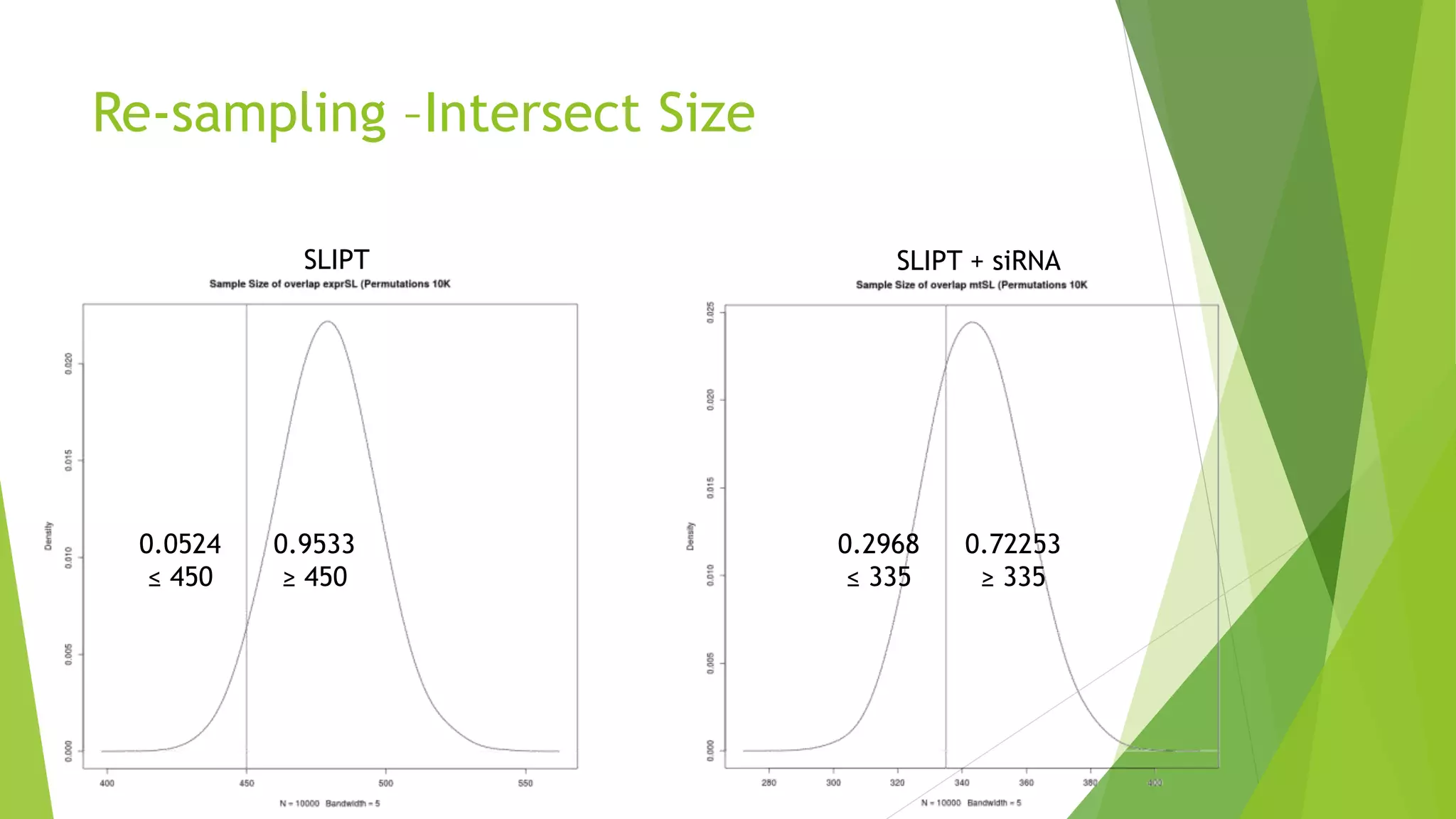
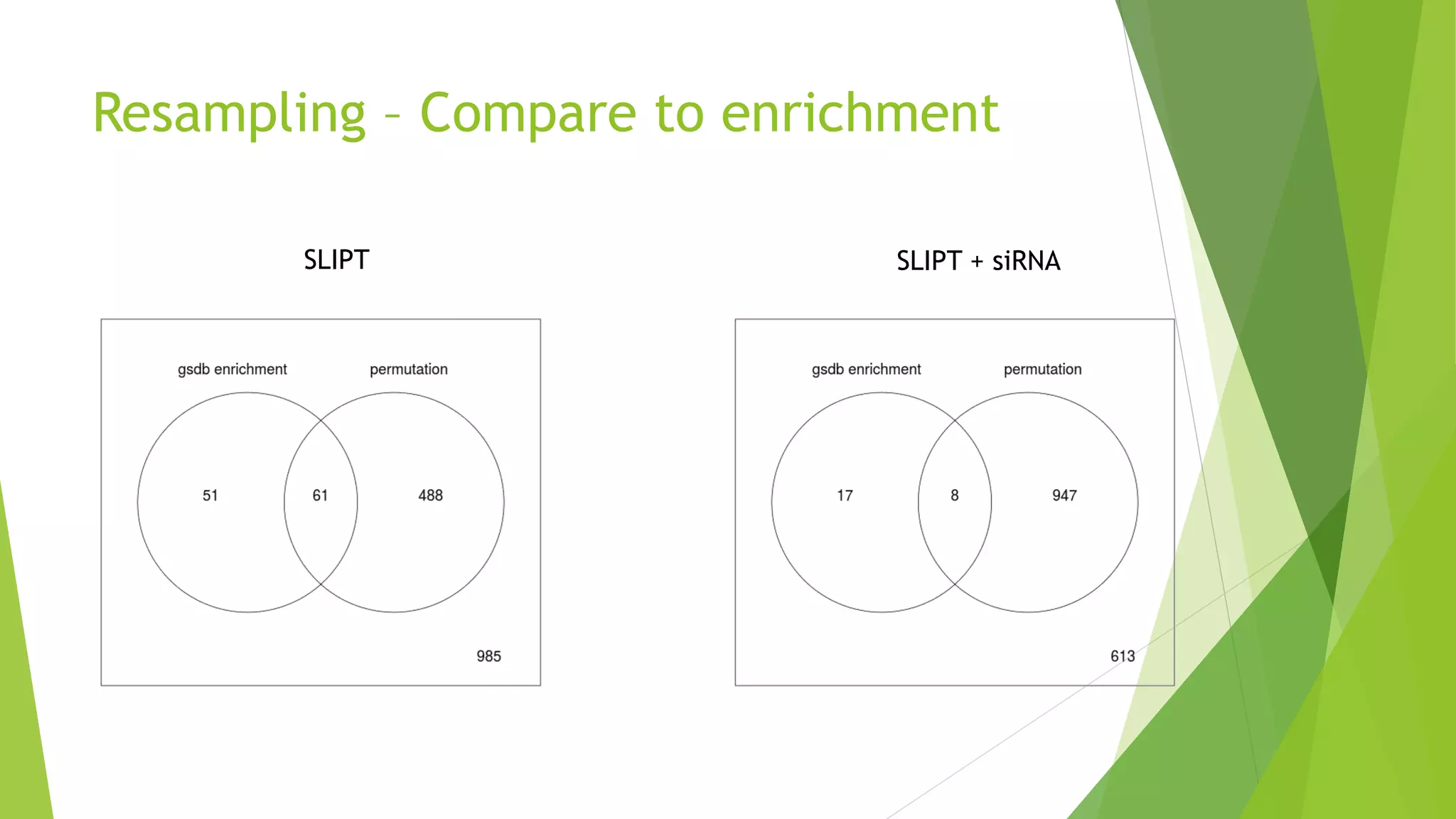
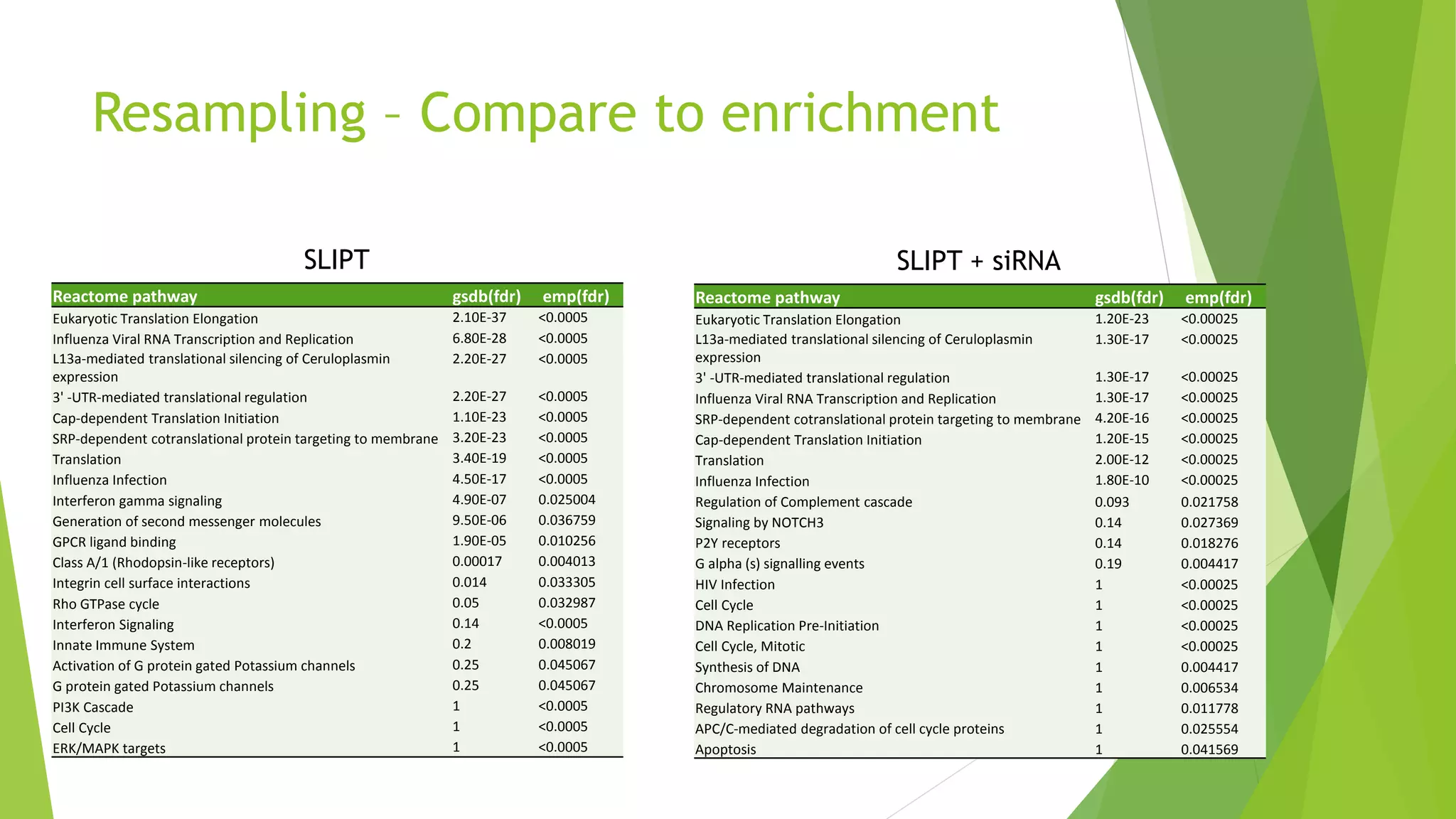


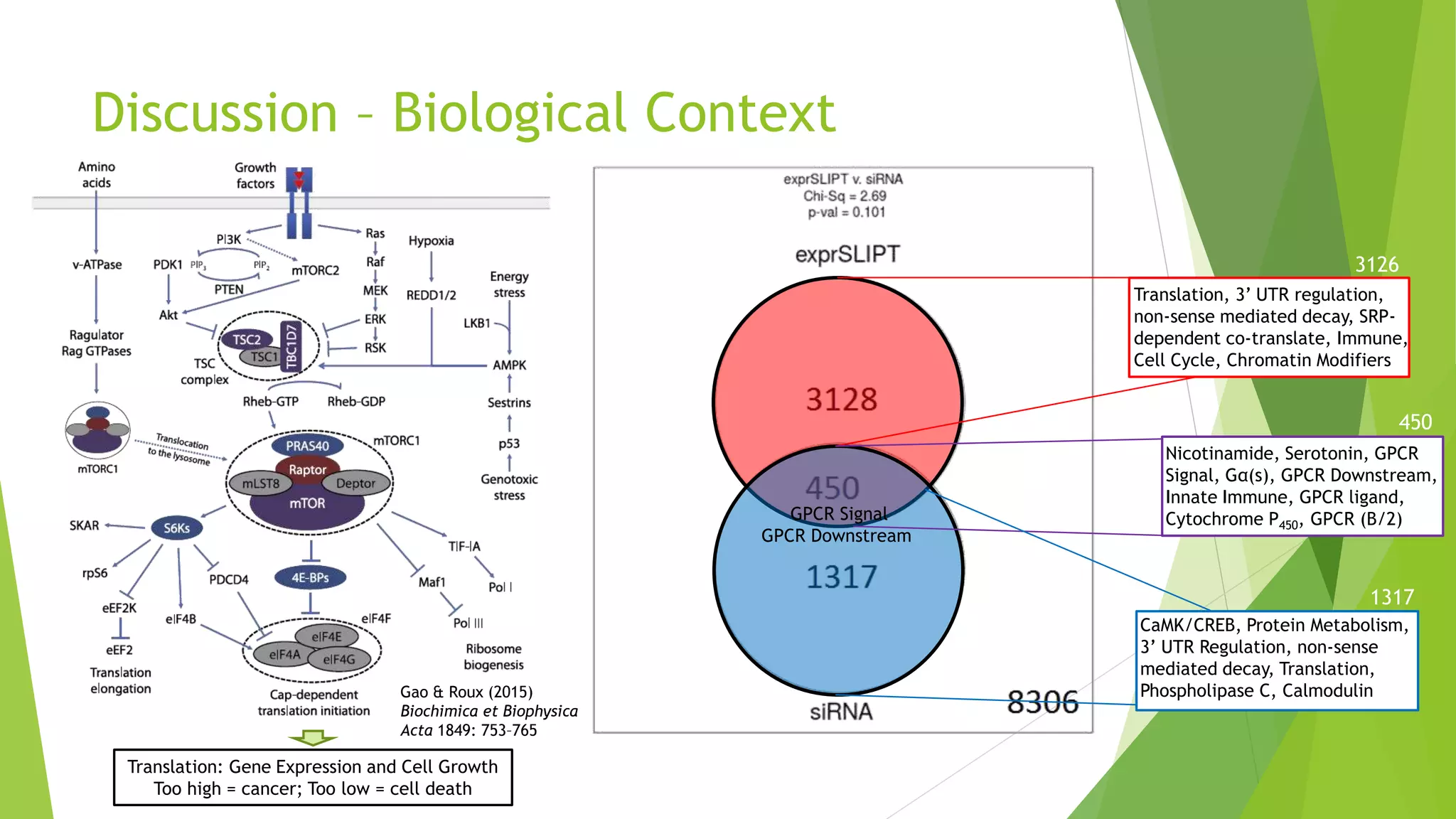






This document summarizes a bioinformatics analysis that used resampling techniques to compare predicted synthetic lethal gene interactions to experimental screening data. The analysis predicted synthetic lethal partners for the CDH1 gene in breast cancer using a method called SLIPT. Pathway enrichment analysis found several pathways enriched in both the SLIPT predictions and intersections with experimental screens, including cell cycle, DNA repair, and WNT signaling pathways. Resampling by permutation was used to generate a null distribution of pathway enrichments and test if overlaps between SLIPT predictions and screens were higher than expected by chance. Several pathways, including translation, nonsense mediated decay, and immune pathways, were significantly enriched in both datasets after multiple testing correction. The analysis provides computational validation of predicted synthetic lethal


























































































