Download to read offline




















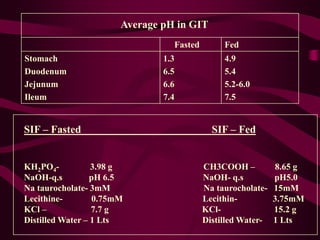


























This document discusses dissolution testing, which is used to evaluate how quickly an active pharmaceutical ingredient is released from its solid dosage form after administration. Key points include: - Dissolution is the process by which a solid enters solution and is controlled by the affinity between the solid and solvent. - Dissolution testing seeks to relate in vitro dissolution to in vivo drug absorption and bioavailability. - The Biopharmaceutics Classification System categorizes drugs based on their solubility and permeability properties to determine the rate-limiting step of absorption. - Procedures for dissolution testing must account for factors like pH, surfactants, and apparatus to mimic conditions in the gastrointestinal tract. - Diss





















































