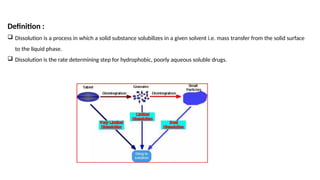
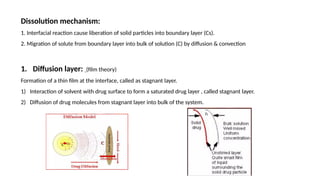
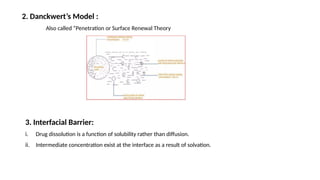
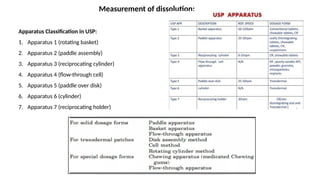
Dissolution is the process of a solid substance being solubilized by a solvent, essential for the bioavailability of hydrophobic and poorly aqueous soluble drugs. Proper dissolution studies are crucial for demonstrating drug release consistency and establishing specifications based on solubility and permeability classifications. Methods for conducting these studies include using different apparatus and media, with specific requirements set for various classes of drugs to ensure quality and performance.