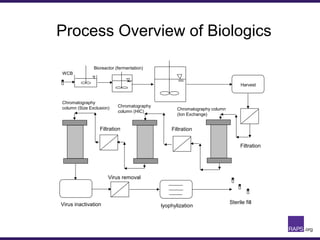
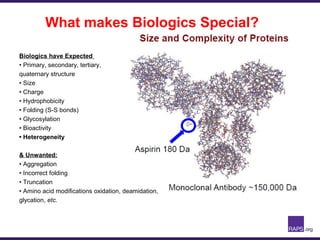
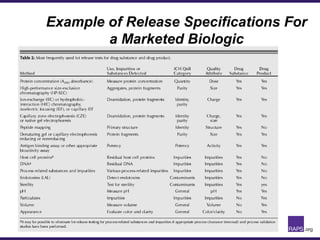
The document discusses the regulatory journey of biologics, focusing on CMC (Chemistry, Manufacturing, and Controls) requirements from the FDA and the challenges of safety, potency, and impurity profiles during the development process. It outlines the importance of effective product characterization, assay methods, and specifications while emphasizing the need for robust viral safety measures and managing process-related impurities. Additionally, the document underscores the necessity of involving regulatory agencies early and throughout the development process to ensure successful compliance.












![List All Actual/Potential Impurities!
• Process-related impurities
Cell-substrate (DNA, HCP, proteases, endotoxins)
Cell-culture (cell-substrate [DNA, HCP, protease];
endotoxin; media components – antibiotics [tetracycline,
gentamicin], hormones [insulin, IGF-1, transferrin],
serum)
Purification (enzymes [DNase/RNase]; resin leachates;
surfactants; residual cleaning agents]
Product-related impurities
FDA Guidance for Review Staff and Sponsors: Gene Therapy 2004](https://image.slidesharecdn.com/cmcbiologicspathwaydraft8-140520180002-phpapp02/85/The-CMC-Journey-in-the-Regulation-of-Biologics-16-320.jpg)



































![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)























![Biotech2007[1]](https://cdn.slidesharecdn.com/ss_thumbnails/biotech20071-12731379674691-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)





















