This document provides an overview of clinical trials, including:
- Definitions of clinical trials and their importance in testing medical treatments.
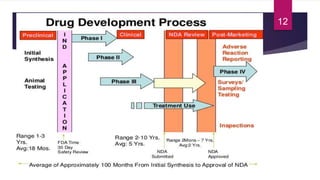
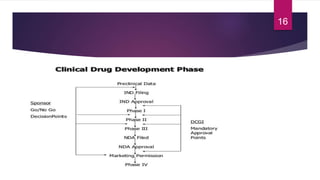
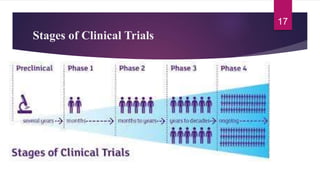
- The various phases of clinical trials (Phases 0-IV) and their objectives in evaluating safety, efficacy, and effectiveness.
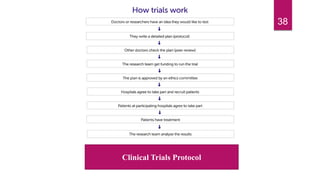
- The roles of institutional review boards, peer review, and regulatory approval in the clinical trial process and new drug application.




















































![roles and responsibilities of Investigator[663]](https://cdn.slidesharecdn.com/ss_thumbnails/investigator663-210616055819-thumbnail.jpg?width=640&height=640&fit=bounds)






























































