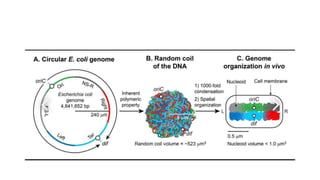
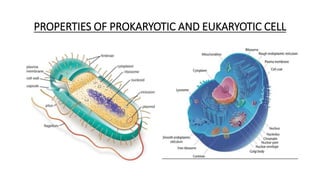
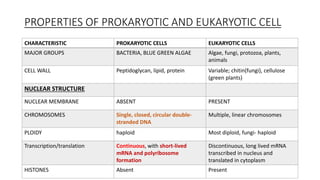
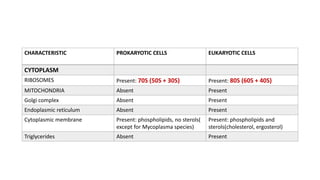
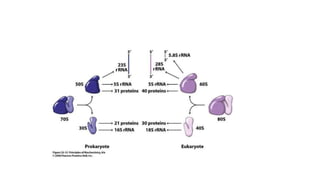
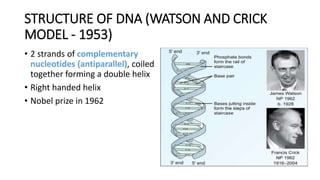
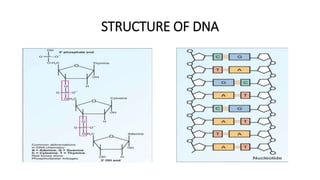
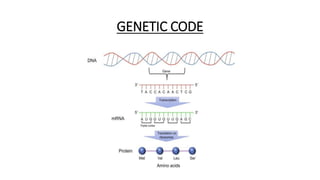
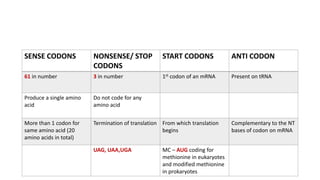
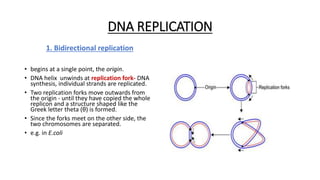
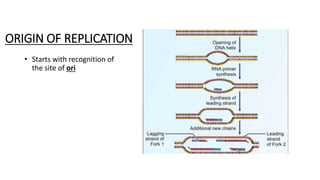
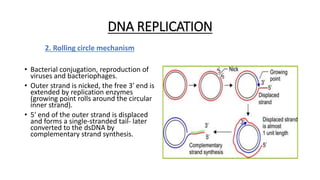
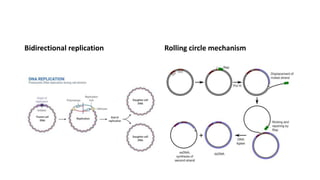
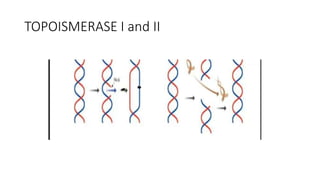
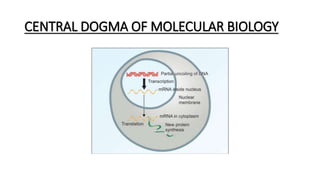
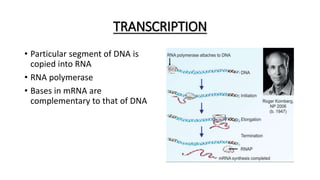
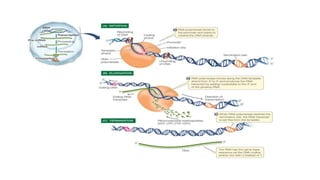
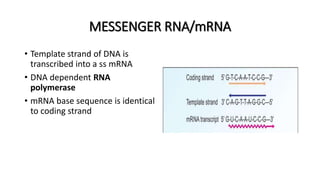
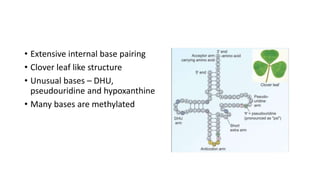
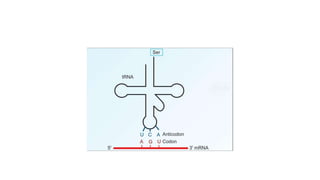
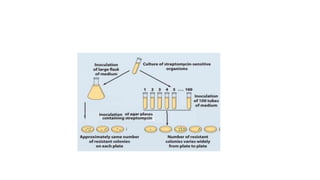
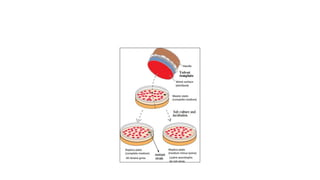
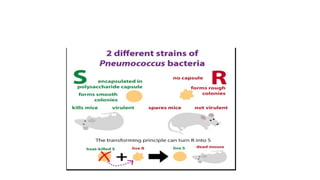
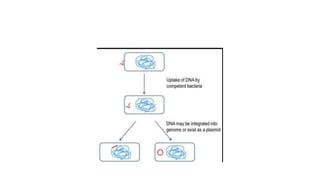
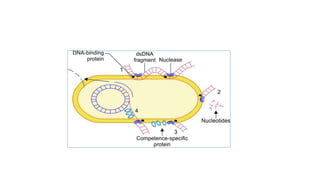
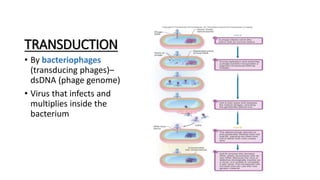
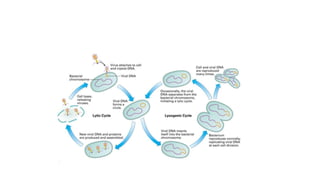
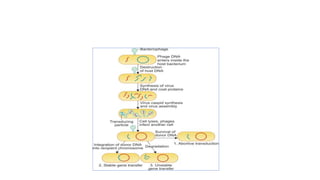
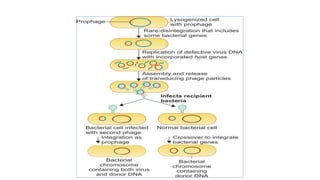
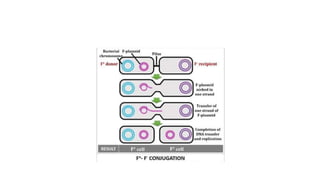
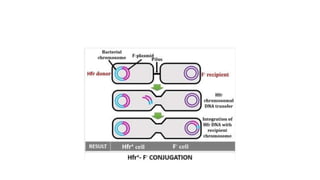
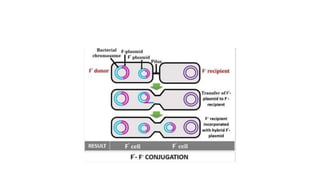
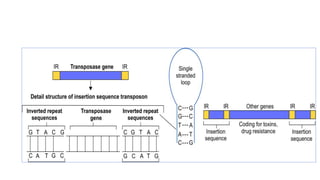
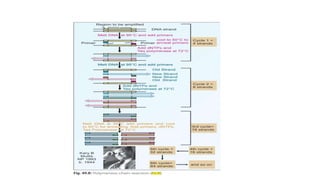
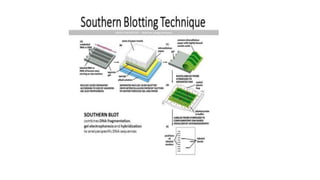
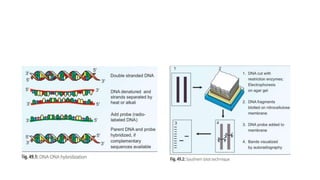
This document provides information on bacterial genetics and DNA structure and replication. It discusses that bacteria typically have a single circular chromosome in the nucleoid region of the cell. Bacterial DNA is usually between 300-1400um in length and comprises around 2-3% of the cell's dry weight. The smallest bacterial chromosome is around 600kb while the largest is around 10mb. Bacterial DNA replication occurs via a bidirectional mechanism using enzymes like helicase, topoisomerase, primase and DNA polymerase. The central dogma of molecular biology involving transcription of DNA to mRNA and translation of mRNA to proteins is also summarized. Key concepts like the lac operon and gene regulation in bacteria are briefly explained.





















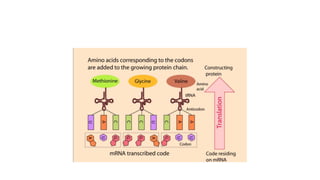
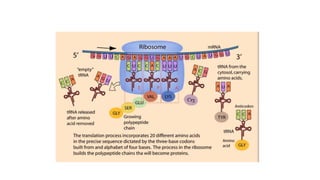

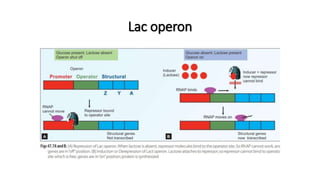




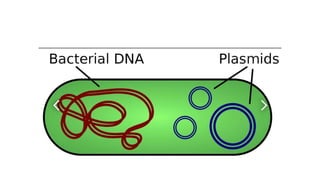


































































































![[K2] DNA GENE EXPRESSION 2019.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/k2dnageneexpression2019-230227101257-d95693f7-thumbnail.jpg?width=640&height=640&fit=bounds)





![Proteinsynthesis [Autosaved]11111111.ppt](https://cdn.slidesharecdn.com/ss_thumbnails/proteinsynthesisautosaved-250216012443-049610aa-thumbnail.jpg?width=640&height=640&fit=bounds)






























