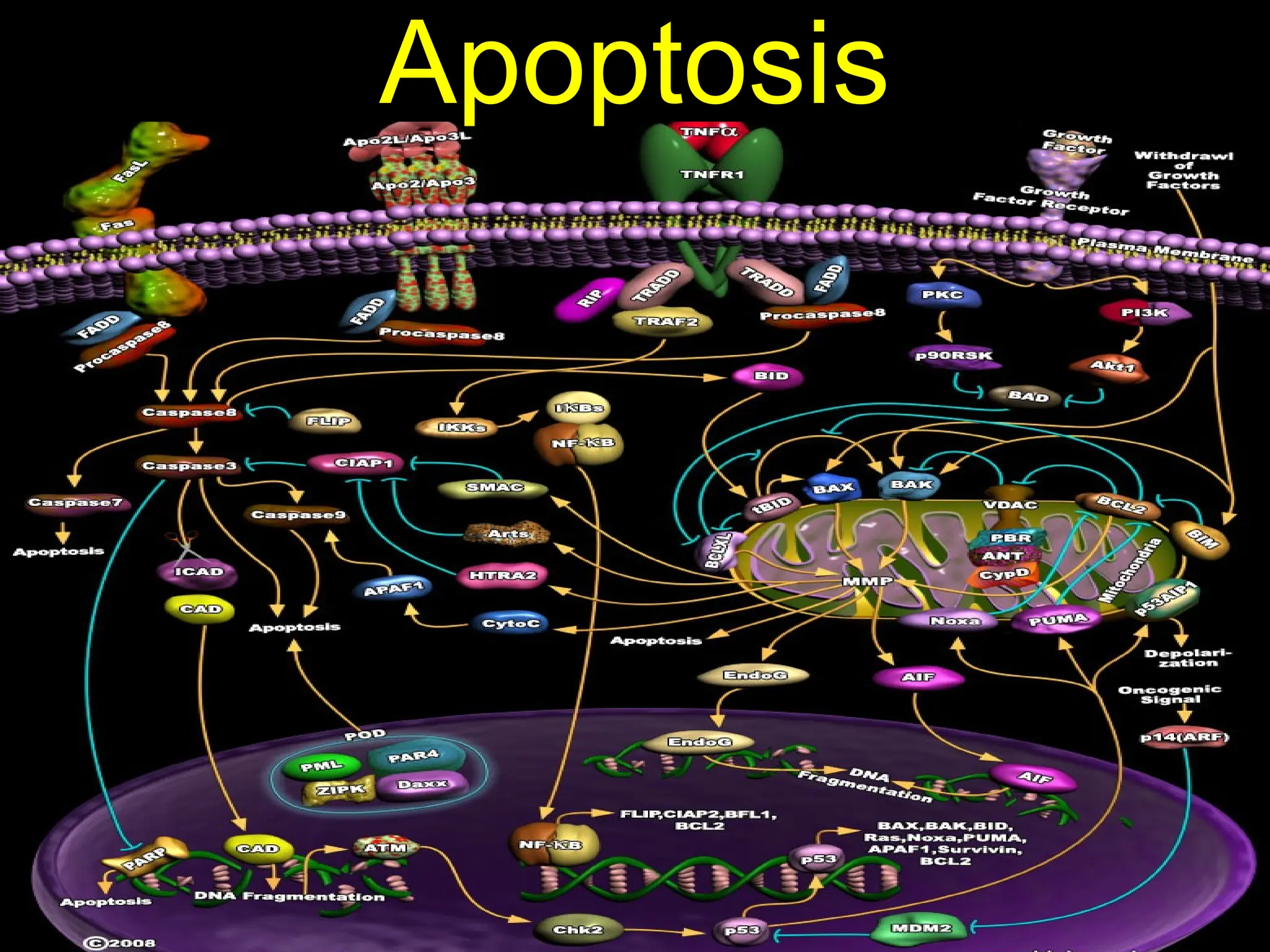
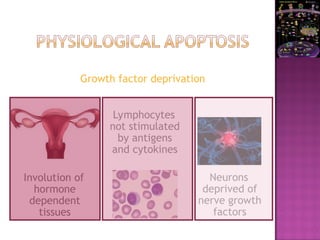
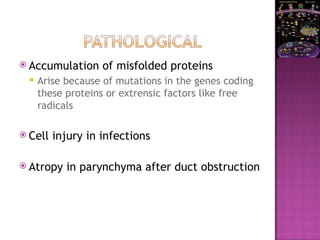
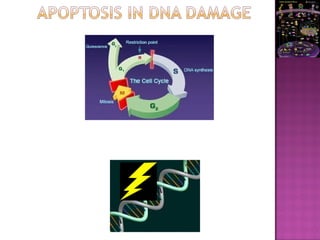
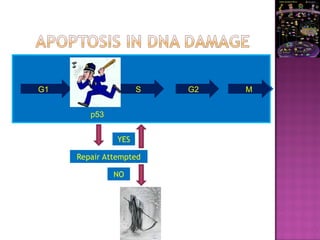
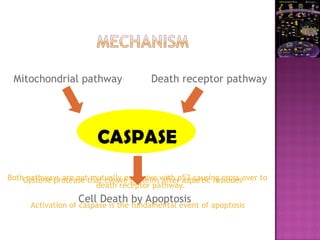
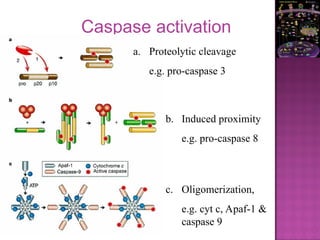
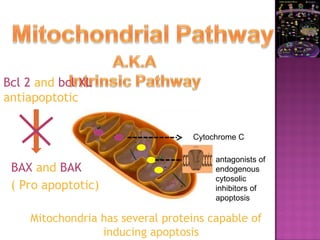
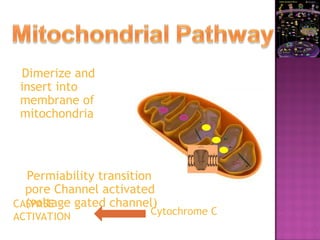
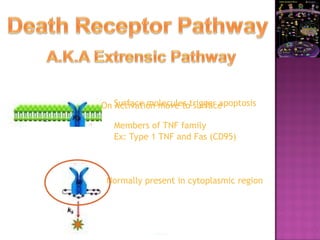
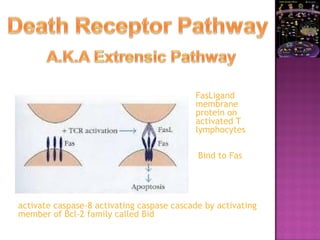
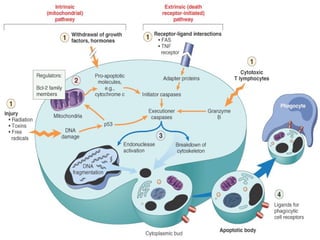
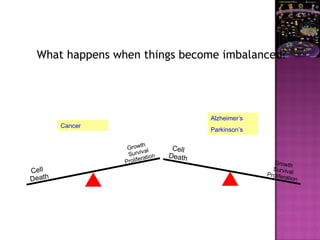
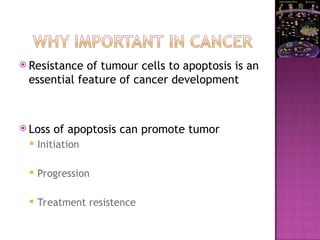
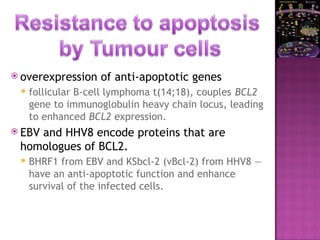
The document discusses apoptosis, a programmed cell death process crucial for maintaining cellular balance and eliminating harmful or unnecessary cells. It details the mechanisms of apoptosis, including pathways involving caspases and Bcl-2 family proteins, and highlights the implications of impaired apoptosis in cancer development and treatment resistance. Additionally, it addresses the challenges in targeting apoptotic pathways for cancer therapy and the dual role of apoptosis in tumor progression and response to chemotherapy.