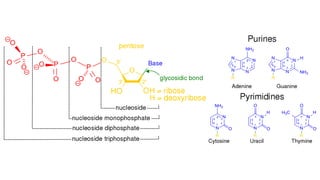
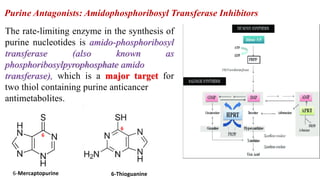
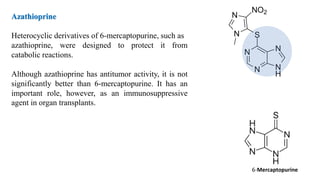
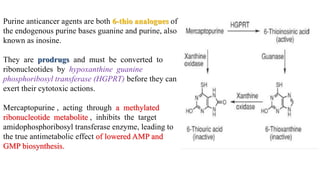
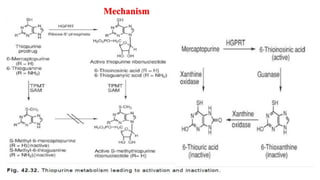
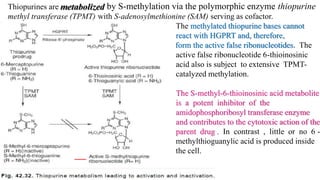
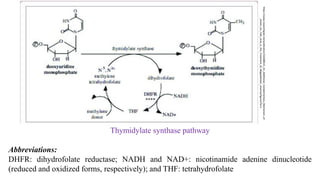
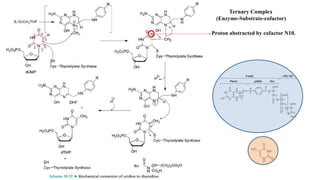
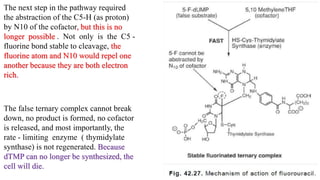
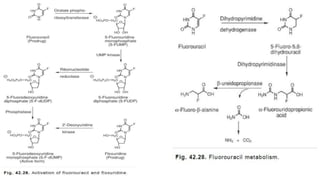
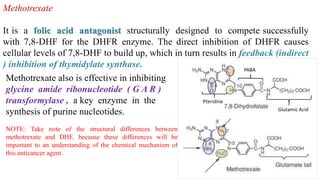
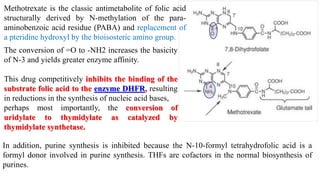
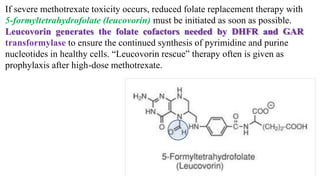
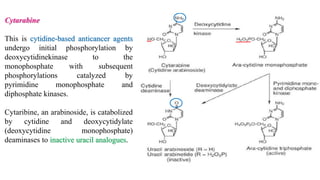
The document discusses various antimetabolites, which are compounds closely related to cellular precursor molecules and act as effective cancer chemotherapy agents by interacting with nucleic acid biosynthesis. Key antimetabolites include mercaptopurine, thioguanine, fluorouracil, and methotrexate, each with distinct mechanisms of action that target critical enzymes in the synthesis of purines and pyrimidines. The document also outlines the therapeutic uses of these agents in treating different types of leukemia and the potential side effects associated with their use.