


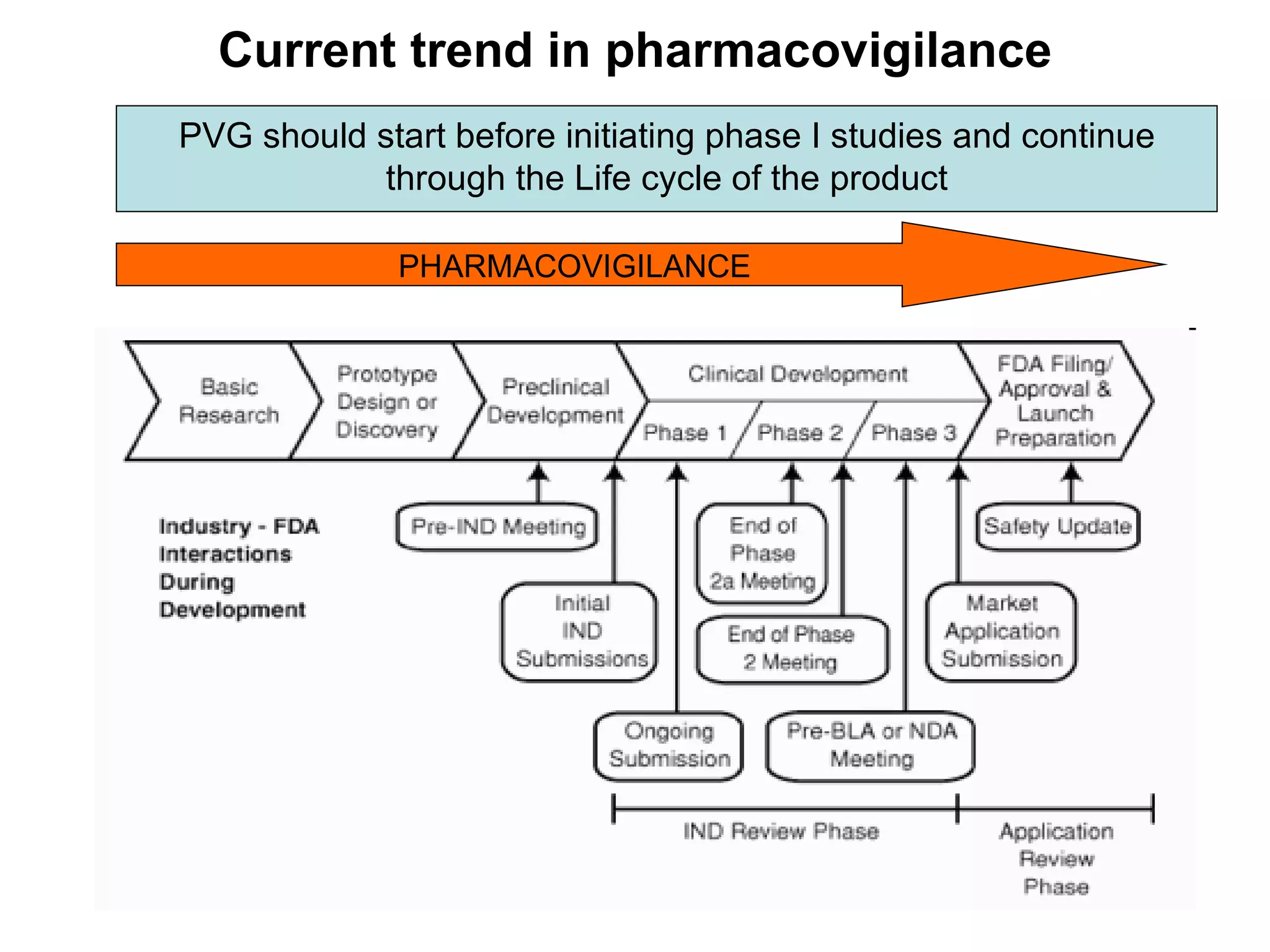



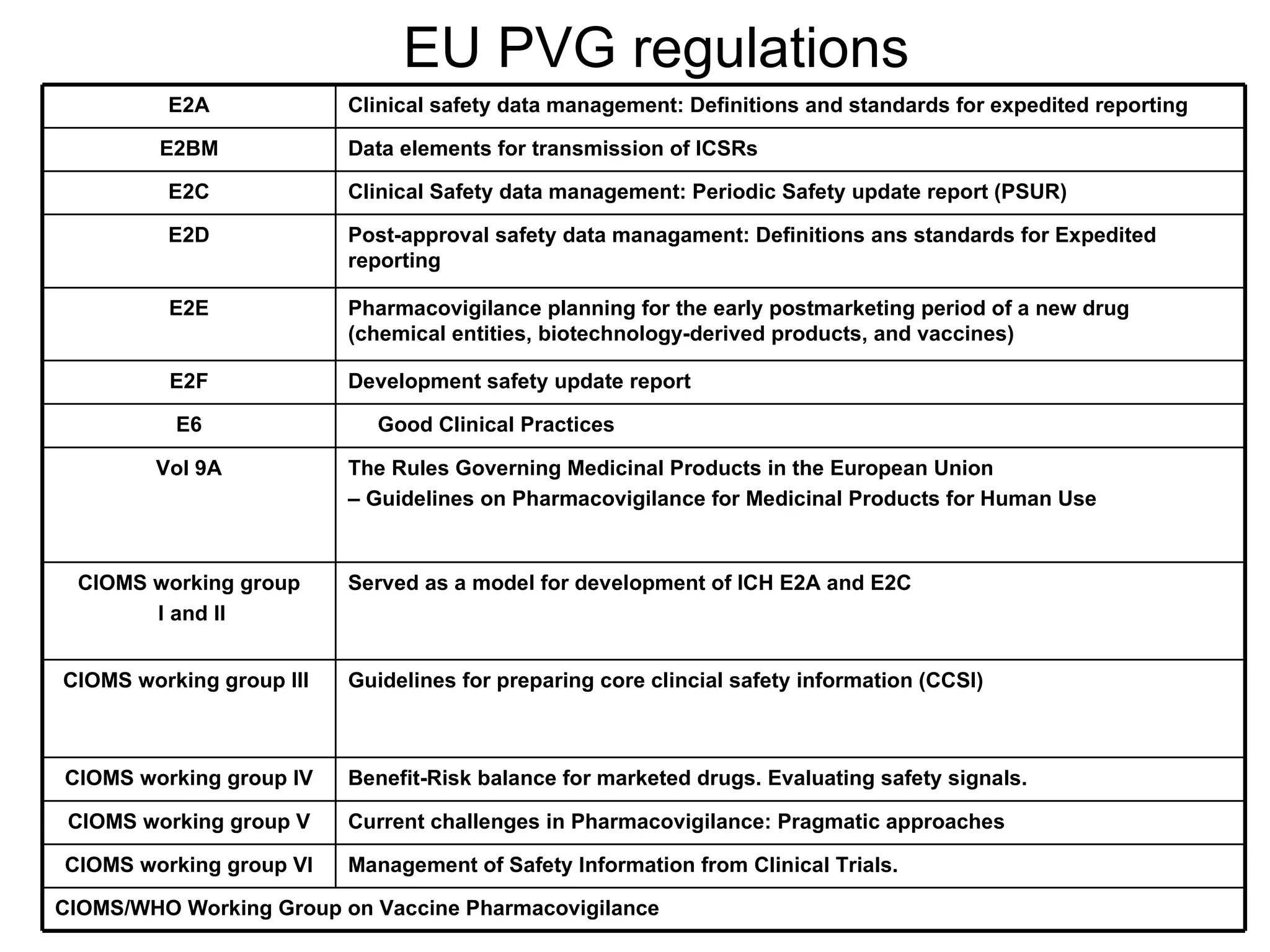

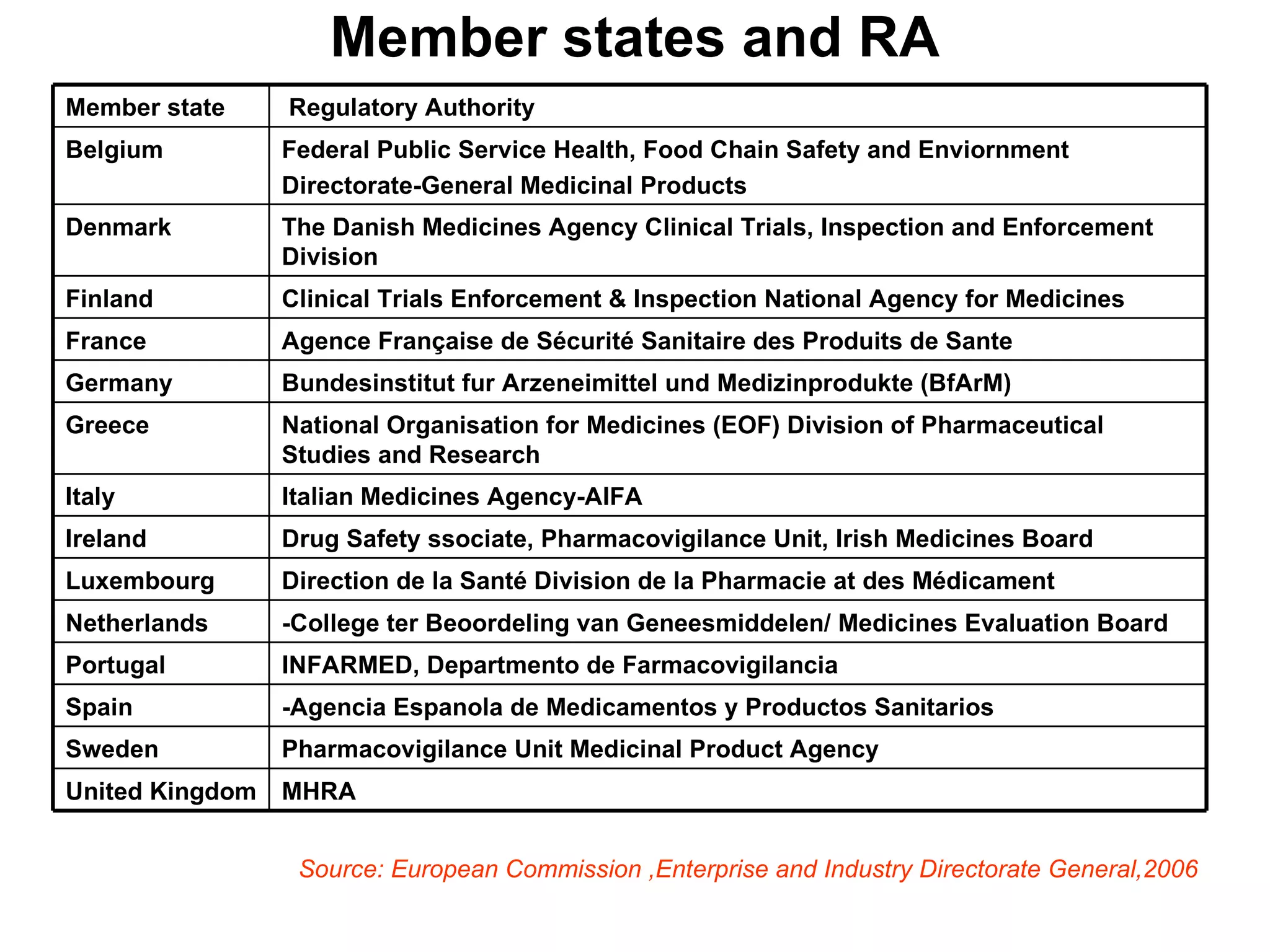





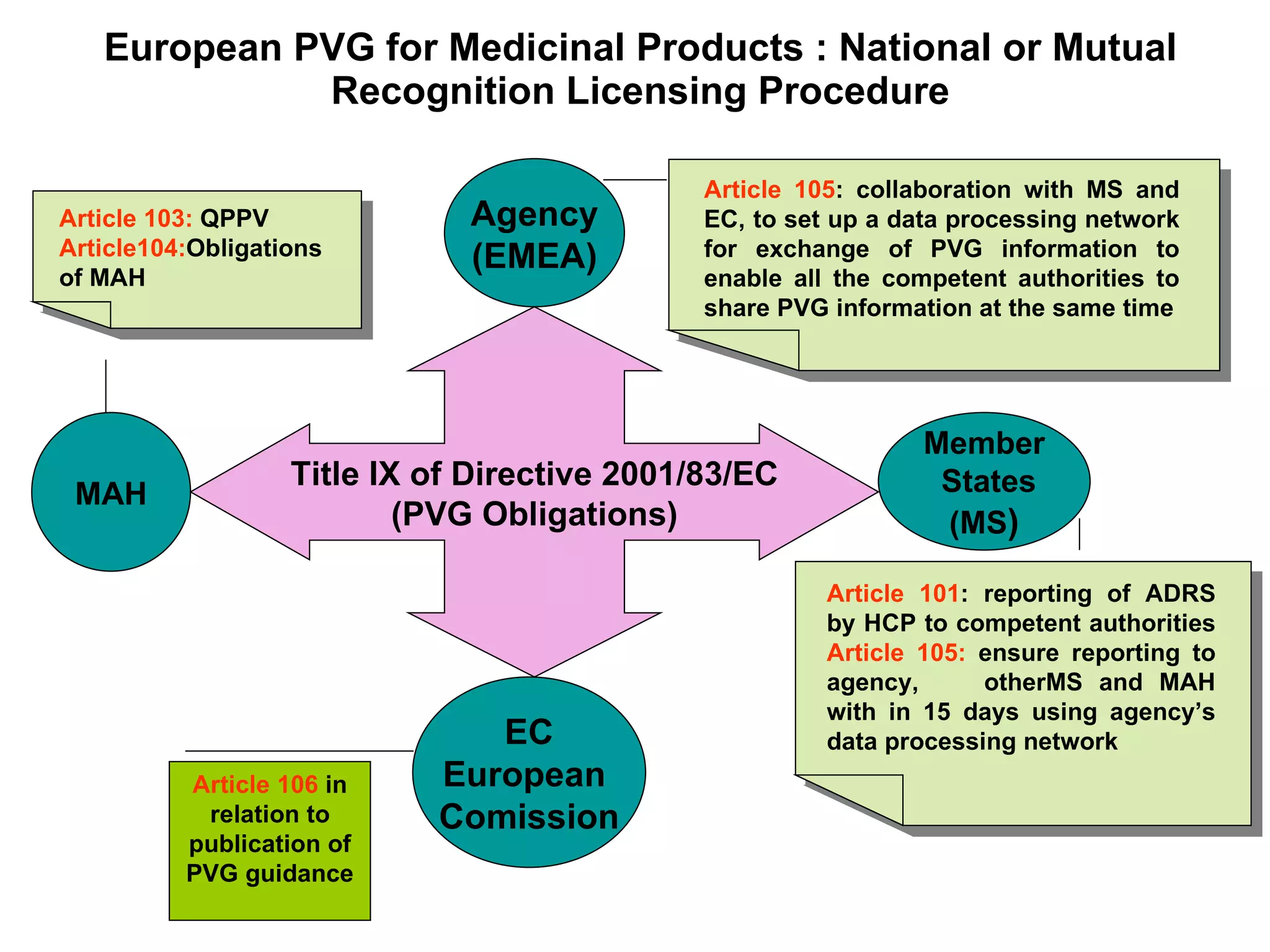












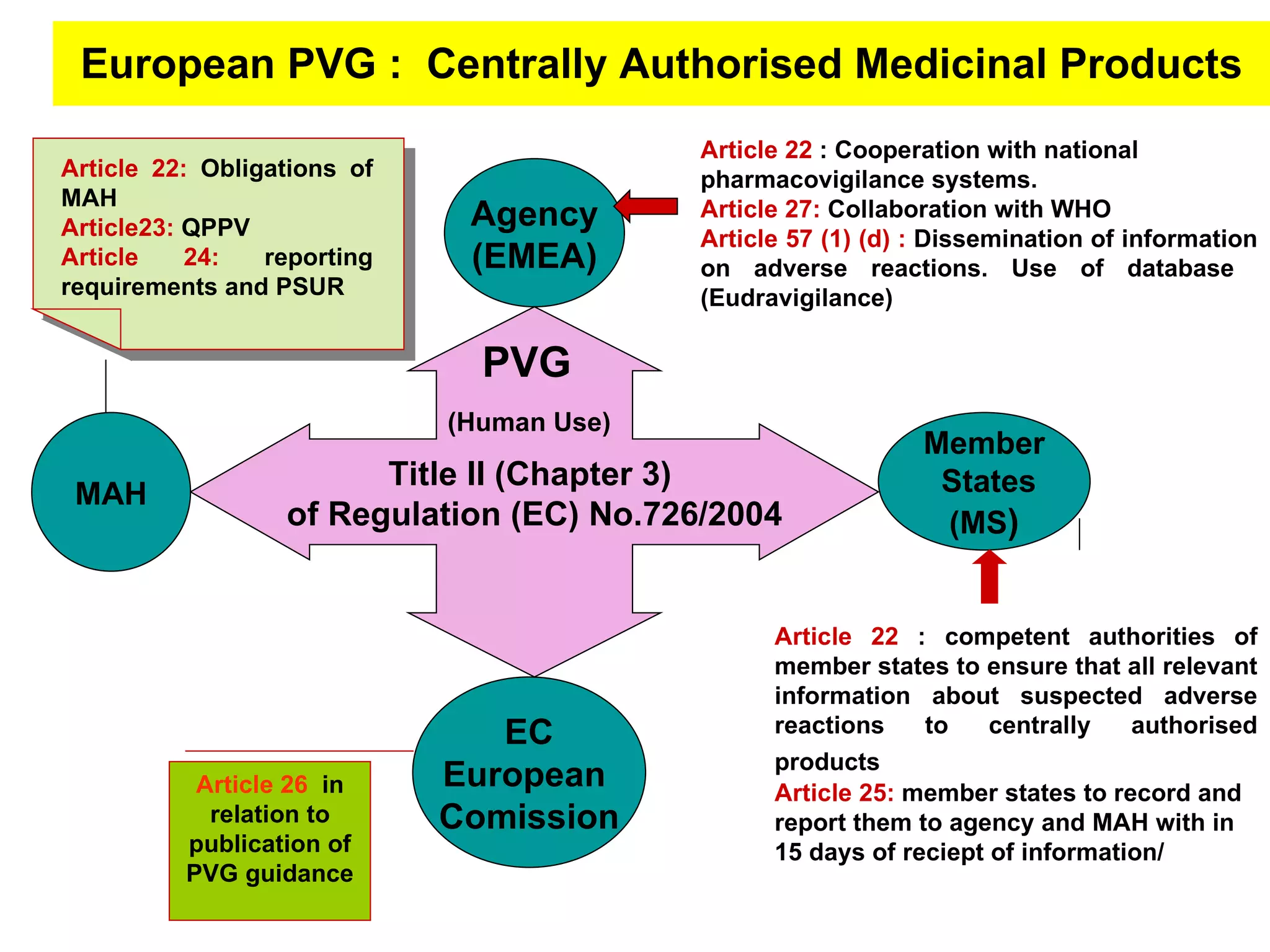








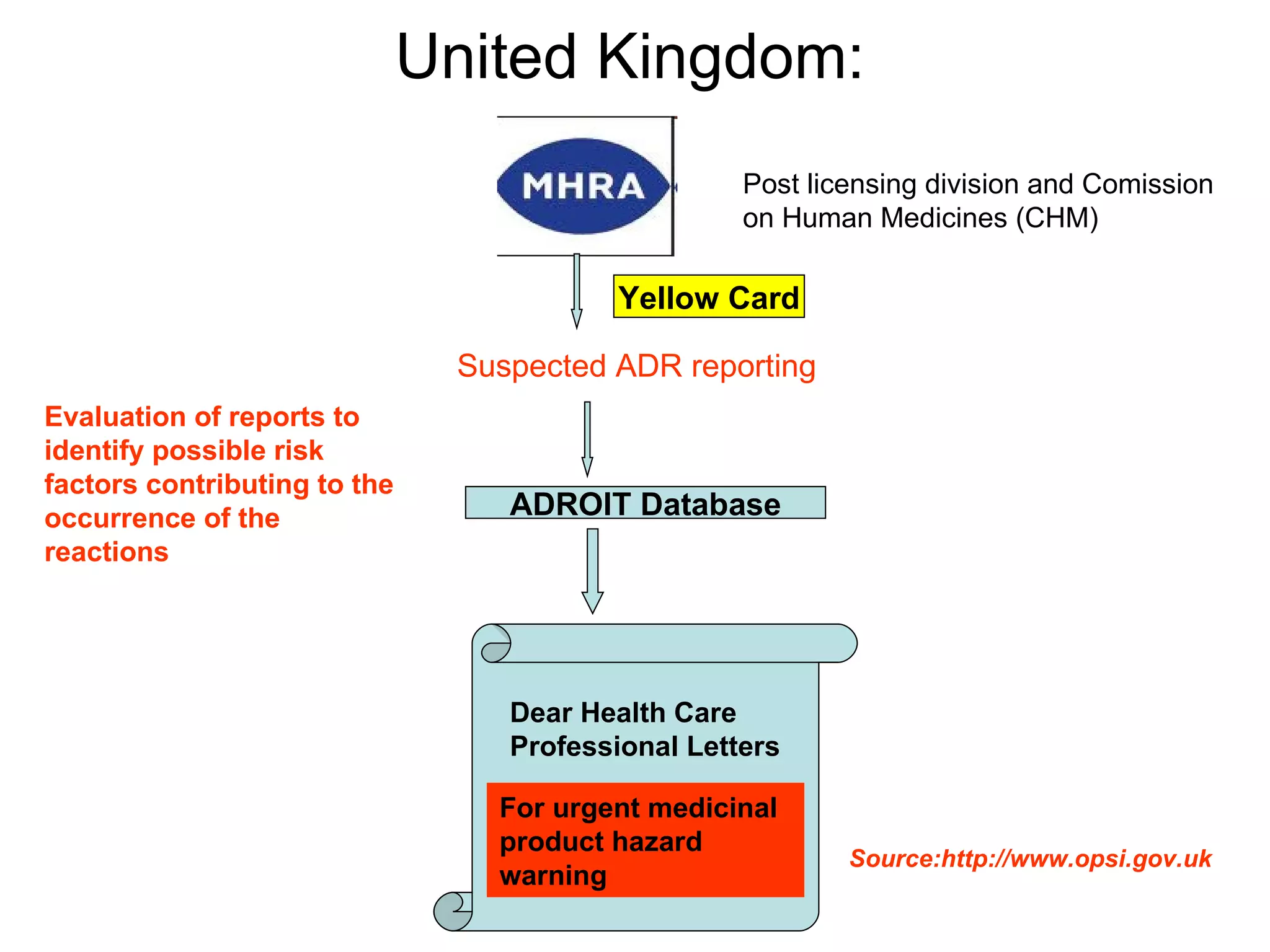








This document summarizes the key principles and regulations of pharmacovigilance in the European Union. It discusses the basis and definitions of pharmacovigilance, the major milestones in EU pharmacovigilance legislation, and the roles and responsibilities of the European Medicines Agency, national competent authorities, marketing authorization holders, and other stakeholders in monitoring the safety of medicines in both pre- and post-authorization phases.


















































![Volume 9 A Guidelines On Pharmacovigilance[1]](https://cdn.slidesharecdn.com/ss_thumbnails/volume9aguidelinesonpharmacovigilance1-12816046179281-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)















































