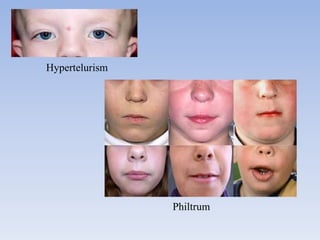
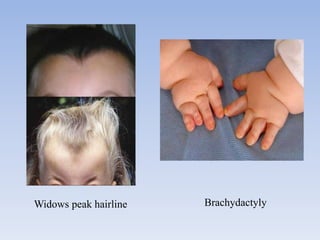
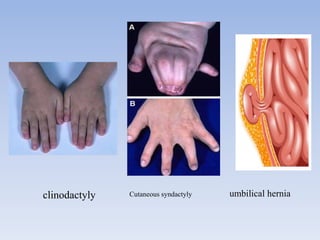
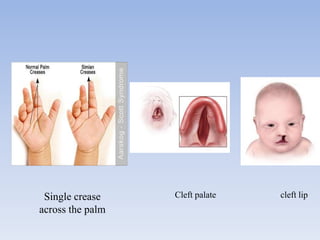
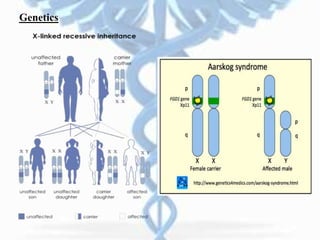
- Aarskog-Scott Syndrome is a genetic disorder caused by mutations in the FGD1 gene, affecting development and causing distinctive facial features and limb abnormalities.
- It mainly affects males and causes short stature, hand abnormalities like short fingers and single palm creases, and genital abnormalities like shawl scrotum.
- There is no cure, but treatment focuses on improving quality of life through procedures to correct physical abnormalities, growth hormone therapy, education support, and genetic counseling.