Download to read offline
![A DFT analysis of the vibrational spectra of nitrobenzene
John Clarkson*, W. Ewen Smith
Department of Pure and Applied Chemistry, Strathclyde University, Thomas Graham Building, 295 Cathedral Street,
Glasgow G1 1XL, Scotland, UK
Received 13 March 2003; accepted 23 April 2003
Abstract
Raman and FTIR, spectra of nitrobenzene, nb, and its isotopomers, nb-15
N, nb-13
C6 and nb-d5; were obtained and the
fundamental vibrational modes assigned with the aid of a B3LYP/6-311 þ G** calculation, without the need for scaling of the
force constants. The changes in vibrational coupling between the nitro and benzene groups upon certain isotopic substitutions
are well modelled by the calculation, which is able to reproduce the isotopic shifts in frequencies for the nitro vibrations, as well
as changes in IR intensities.
q 2003 Elsevier B.V. All rights reserved.
Keywords: Density functional theory; Isotopomers; Vibrational analysis
1. Introduction
Nitrobenzene, the simplest nitro aromatic com-
pound, is the parent molecule of a range of important
compounds, including explosives such as 2,4,6-
trinitrotoluene. It is also related to nitrophenols,
including the biologically important nitrotyrosine,
whose presence in proteins is often the result of
damage by peroxynitrite [1–3]. A full theoretical
understanding of the molecular properties of nitro-
benzene can therefore inform on the properties of
these other larger molecules.
This paper focuses on the use of modern density
functional theory (DFT), to fully account for the
experimental vibrational Raman and IR data from
nitrobenzene and its isotopomers. The vibrational
spectroscopy of nitrobenzene has been the subject of
numerous studies [4–15]. The exact nature of the
nitro vibrational modes, however, had been either
vague or contradictory. A thorough analysis of the
vibrational frequencies of the C–NO2 moiety on the
basis of a HF/6-31G* calculation, where the force
constants had been scaled, was published by Shlya-
pochnikov et al. [15]. This appeared to provide a clear
assignment of the nitro bands and seemed to fully
account for the isotopic shifts that occur for the
various isotopomers of nitrobenzene.
A vibrational analysis of 2-nitrophenol making use
of a B3LYP/6-31G* calculation also had to scale the
force constants to obtain a satisfactory fit of the
frequencies to the experimental data [16]. The scaling
0022-2860/03/$ - see front matter q 2003 Elsevier B.V. All rights reserved.
doi:10.1016/S0022-2860(03)00316-8
Journal of Molecular Structure 655 (2003) 413–422
www.elsevier.com/locate/molstruc
* Corresponding author. Tel.: þ44-141-552-4400; fax: þ44-141-
552-0876.
E-mail address: john.clarkson@strath.ac.uk (J. Clarkson).
Abbreviations: nb, nitrobenzene; nb-15
N, 15
N enriched
nitrobenzene; nb-d5, deuterium enriched nitrobenzene; nb-13
C6,
13
C enriched nitrobenzene.](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-1-2048.jpg)
![of force constants for molecules such as nitrobenzene
and 2-nitrophenol was needed as it was thought that a
purely computational approach to the force field or
frequencies for such molecules was not possible.
Systematic calculation errors were compensated for
and good agreement with the observed experimental
frequencies was achieved by using a few empirical
scale factors for the calculated force constants
[15,16].
The present authors reported a recent successful
DFT analysis of the structure and vibrations of 2,4,6-
trinitrotoluene, TNT, using the hybrid density func-
tional B3LYP and the 6-311 þ G** basis set [17],
without the need to scale the force constants or
frequencies. The good fit between the theoretical and
experimental frequencies for TNT was only achieved
by use of the triple zeta, 6-311 þ G** basis set; A
B3LYP calculation of TNT with the double zeta, 6-
31G* basis set gave poor results. Following this, the
authors decided to re-examine the vibrational spectra
of nitrobenzene to test if a similar DFT analysis could
successfully model the vibrational spectra of nitro-
benzene and account for the isotopic shifts without the
need to rescale the force constants.
We present FTIR and Raman data for liquid
nitrobenzene, nb, nb-d5; nb-15
N, and nb-13
C6 and
assign the spectra on the basis of a B3LYP/6-
311 þ G** calculation. The changes in the
vibrational mode coupling between the nitro group
and the benzene ring for the various isotopomers are
well modelled by the B3LYP/6-311 þ G** calcu-
lation and previous assignments of the vibrational
spectra are revised. The present analysis represents a
reappraisal of the analysis of Shlyapochnikov et al.
[15], presenting new vibrational data for nb-13
C6 and
new insights into the coupling of the nitro and
benzene modes.
2. Methods
Nitrobenzene and d5-nitrobenzene were obtained
from Aldrich. 15
N (98%) enriched nitrobenzene and
13
C6 (99%) enriched nitrobenzene, were obtained
from Cambridge Isotope Laboratories.
Raman spectra were obtained using a Renishaw
micro-Raman system 1000 spectrometer with 785 nm
excitation. FTIR spectra were obtained using a
Nicolet Impact 400D.
Geometry optimisation and frequency analysis of
nitrobenzene was performed using GAUSSIAN 98 [18]
with the B3LYP hybrid density functional [19,20]
using the 6-31G* [21–23] and 6-311 þ G** [24,25]
basis sets.
3. Results and discussion
Computed bond lengths and angles for nitroben-
zene together with the electron diffraction and
microwave data (Fig. 1) are shown in Table 1.
Previous MP2 calculations showed the C–C bonds as
getting longer the farther away from the NO2 group
attachment point, in contrast to the HF calculation
[15]. The present B3LYP calculations is in agreement
with the HF calculation in that only the farthest away
CC bond length is notably longer, the other two being
near equal. There is a large difference in the CN and
NO bond lengths modelled by the B3LYP calculations
compared to previous HF and MP2 calculations. The
improvement in the MP2 NO2 bond lengths over the
HF values is in increasing the CN bond length,
however, it overestimates the NO bond length [15].
The B3LYP calculations increase further the CN bond
length, but decreases the NO bond length. The effect
of the 6-311 þ G** basis set on the B3LYP
functional, compared to the B3LYP/6-31G* structure
Fig. 1. Labelling of the atoms of nitrobenzene.
J. Clarkson, W. Ewen Smith / Journal of Molecular Structure 655 (2003) 413–422414](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-2-2048.jpg)
![is to further increase the length of the CN bond and
decrease the NO bond length.
Raman and FTIR spectra for nb, nb-15
N, nb-13
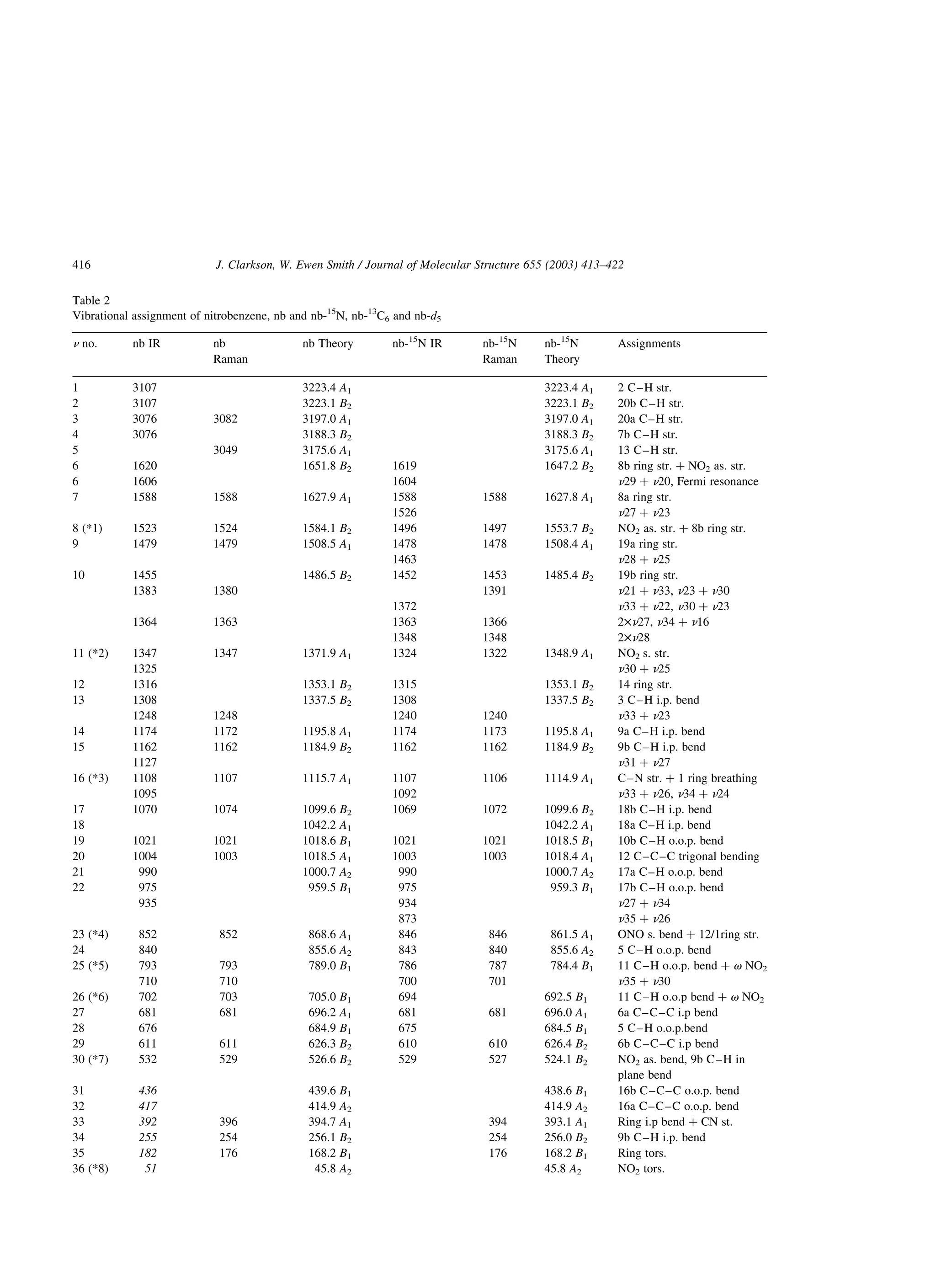
C6
and nb-d5 are shown on Fig. 2 and assignments based
on the B3LYP/6-311 þ G** calculation are presented
on Table 2. The calculated B3LYP/6-31G* frequen-
cies, the IR intensities and their changes for the
various isotopomers of nitrobenzene were found to be
inaccurate. A similar result was found in the TNT
study [17]. The assignments on Table 2 are based on a
visual inspection of the calculated eigenvectors and
the benzene ring modes are numbered after the Wilson
[26] notation as recommended by Varsanyi [27].
The C2v symmetry of nitrobenzene classifies the
normal vibrations into 13 A1, 12 B2, 7 B1 and 4 A2
modes. Following the analysis of Shlyapochinkov
et al. [15], seven of these modes are identified with the
C–NO2 moiety; three A1, (ns NO2, n CN and d ONO),
two B2, (nas NO2 and t NO2), one B1 (v NO2), and
one A2 (x NO2) modes. Eight NO2 modes were
selected by Shlyapochinkov et al. [15] due to the fact
that the v NO2 out-of-plane deformation takes part
nearly to the same extent in both n25 and n26; *5 and
*6, respectively. The greater isotopic shift for *6 in
nb-15
N, Table 3, suggests this fundamental as
predominantly the NO2 out of plane deformation
mode. The present analysis, however, shows that this
Table 1
Computed B3LYP/6-311 þ G** bond lengths and angles for nitrobenzene
HF 6-31G*a
MP2 6-31G*a
MP2 6-31G**a
B3LYP 6-31G* B3LYP 6-311 þ G** Microwavea
C1–C3 1.3833 1.3911 1.3905 1.3936 1.3915 1.3748
C3–C4 1.3833 1.3934 1.3928 1.3934 1.3914 1.4026
C2–C4 1.3867 1.3960 1.3953 1.3976 1.3952 1.3958
C–N 1.4588 1.4700 1.4699 1.4734 1.4807 1.4916
N–O 1.1938 1.2413 1.2412 1.2308 1.2243 1.2272
C3–C1–C5 122.32 122.77 122.76 122.35 122.28 124.99
C1–C3–C4 118.51 118.11 118.12 118.47 118.51 117.11
C2–C4–C3 120.10 120.33 120.50 120.19 120.20 120.30
C4–C2–C6 120.46 120.01 120.01 120.33 120.31 120.18
C–N–O 117.70 117.66 117.62 117.70 117.67 117.82
O–N–O 124.61 124.69 124.77 124.60 124.65 124.35
C3–H10 1.0709 1.0839 1.0787 1.0829 1.0811 1.080
C4–H9 1.0742 1.0866 1.0812 1.0859 1.0834 1.0829
C2–H8 1.0750 1.0867 1.0813 1.0863 1.0839 1.0803
C1–C3–H10 120.02 119.93 119.80 119.52 119.63 120.7
C2–C4–H9 120.24 120.10 120.07 120.19 120.18 120.05
C4–C2–H8 119.77 120.00 120.00 119.84 119.85 119.91
a
From Ref. [15].
Fig. 2. Raman and FTIR spectra of nitrobenzenes, (A) nb-15
N, solid
line, (B) nb-13
C6, (C) nb-d5. Dotted lines in (A) and (B) are
nitrobenzene, nb spectra.
J. Clarkson, W. Ewen Smith / Journal of Molecular Structure 655 (2003) 413–422 415](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-3-2048.jpg)


![NO2 mode mixes with benzene mode 11, yielding the
two *5 and *6 modes. This NO2 mode also mixes with
benzene mode 11 in nb-d5 giving the largest isotopic
shift in Table 3, due to the large contribution from C–
H/D out of plane bending in these modes. Shlyapo-
chinkov et al. calculate þ19.7 and 213.4 cm21
shifts
for nb-d5 modes *5 and *6, respectively, which
the present analysis suggests are incorrectly assigned
[15]. The present calculation predicts that of all the
isotopomers of nitrobenzene, except for nb-d5, the *6
mode has the highest contribution from the NO2 of-of-
plane deformation mode. It appears that the *5 band of
nb-d5 was mistaken for the nitrobenzene *6 band by
Shlyapochinkov et al. [15].
There is very good agreement between our
calculated isotopic shifts and those observed exper-
imentally, Tables 2 and 3, giving extra confidence in
the modelled vibrations. Table 4 shows our calculated
frequencies for all of the isotopomers of nitrobenzene
studied to date, including nb-18
O16
O, nb-18
O2 and nb-
p-d1.
The asymmetric nitro vibration *1, is calculated
to couple to the benzene mode 8b, (n6 and n8 for nb
and nb-13
C6, Fig. 3). The degree of this mixing
dramatically changes in nb-13
C6, where the highest
non-C–H stretching mod, (n6; Fig. 3) is predicted to
have the second highest IR intensity, Fig. 4. The
benzene 8b mode of nitrobenzene is involved in Fermi
resonance with the combination mode 12 þ 6b,
(1004 þ 611 cm21
), resulting in two IR bands at
1620 and 1606 cm21
. Identical Fermi resonance is
observed for nb-15
N and also for nb-13
C6, were two
strong bands, at 1568 and 1553 cm21
, are observed.
The increased contribution of the nb-13
C6 asymmetric
nitro stretch to the highest non C–H stretching mode,
modelled by the calculation, is shown by the large
increase in IR absorption for these Fermi resonance
bands, Fig. 4.
The 1620 and 1606 cm21
IR bands were correctly
identified as a Fermi resonance pair by Kuewae and
Machida, though the benzene mode combination band
was wrongly identified as 1 þ 6b [13]. These bands,
however, were incorrectly assigned to fundamentals
by Shlyapochinkov et al. [15]. There are many
combinations and overtones present in the Raman
and IR spectra of the nitrobenzene above 1650 cm21
,
which we have not assigned. Combinations and
overtones below 1650 cm21
have been assigned to
fully account for all bands in the fundamental region
of the spectra, Table 2.
Table 3
List of observed and calculated isotopic frequency shifts for the C–NO2 moiety for nitrobenzene. Values in parenthesis calculated by
Shlyapochinkov et al [15].
No. Vibration nb-13
C6
a
nb-15
Na
nb-d5
a
Nb-16
O,18
O [15] Nb-18
O2 [15] Para-d-nb [15]
Obs. Calc. Obs. Calc Obs. Calc. Obs. Calc. Obs. Calc. Obs. Calc.
*1 nas NO2 210 221.0 228 230.4
(227.9)
26 29.4
(213.7)
213 210.8
(25.1)
221 223.7
(220.1)
24 23.5
(26.0)
*2 23 21.1 223 222.9
(225.7)
þ14, 26 þ5.2
(þ7.7), 22.7
214 216.9
(28.3), 220.9
227 235.2
(229.2)
21 0.0
(20.1)
*3 n CN 227 229.3 21 20.8
(20.2)
231 233.3
(240.3)
24 22.3
(22.3)
24.6
(28.8)
0 20.1
(20.1)
*4 d ONO 26 25.8 26 27.1
(26.0)
27 27.1
(23.1)
212 27.2
(27.3)
226 229.1
(28.4)
0 20.7
(20.6)
*5 v NO2 29 28.5 26 24.6
(25.5)
273 265.4
(þ15.5)
0 20.6
(20.4)
22 21.3
(21.6)
230 236.4
(233.9)
*6 23 22.0 28 212.5
(210.8)
290 290.7
(219.7)
0 22.1
(21.0)
22 24.3
(23.7)
214 25.1
(214.2)
*7 t NO2 26 25.5 22 22.5
(22.3)
219 213.8
(213.4)
212 28.2
(24.3)
224 216.5
(216.2)
22 21.2
(21.3)
*8 x NO2 20.6 0.0
(0.0)
21.7
(21.2)
20.8
(20.5)
21.5
(22.0)
0.0
(0.0)
Notation: n; d; v; t; and x-stretching, bending, wagging, rocking and tortional vibrations, respectively; a, as-symmetric and antisymmetric.
a
Observed values from our own data shown on Fig. 2, Table 2.
J. Clarkson, W. Ewen Smith / Journal of Molecular Structure 655 (2003) 413–422 419](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-7-2048.jpg)

![group and the greatest IR absorption, Fig. 4, whereas,
experimentally they are of near equal intensity.
Kuewae and Machida also assign the 1360
and 1341 cm21
features to symmetric nitro modes
and their calculated force field correctly predicts the
positive and negative isotopic shifts [13].
The assignment of *4 for nb-d5 is assigned to the
band at 846 cm21
in agreement with Shlyapochinkov
et al. [15] and Laposa [14]. The intense Raman band
at 872 cm21
, which may be mistaken for *4, is
assigned to a C–D in plane bending mode, with a very
small contribution from ONO bending.
Interestingly the calculation predicts that the
reduction in molecular symmetry to Cs upon substi-
tution of one of the nitro group oxygen atoms with
18
O, nb-16
O18
O, results in the symmetric nitro stretch
Fig. 3. The B3LYP/6-311 þ G** eigenvectors for selected nitro
modes of nitrobenzene isotopomers. The differing lengths of arrow
show relative differences in amplitude of the vibrating atom.
Fig. 4. Comparison of the predicted B3LYP/6-311 þ G** and
experimental IR data for nb, nb-14
N, nb-13
C6 and nb-d5.
Fig. 5. Raman and IR spectra for nb-d5 between 1390 and
1310 cm21
; the two largest fitted curves are assigned to nitro
symmetric stretch modes, n9 and n11; coupled to benzene mode
19a.
J. Clarkson, W. Ewen Smith / Journal of Molecular Structure 655 (2003) 413–422 421](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-9-2048.jpg)
![mixing with benzene mode 14, Fig. 3. This gives rise
to two symmetric nitro stretch modes, *2, that are
predicted to have near identical IR intensity, near half
of that for the *2 mode of nb, separated by 4 cm21
.
The experimental data only shows one intense IR
band for 16
O18
O-nb [10], however, symmetric nitro IR
bands are generally broad features and any possible
splitting of this band may be obscured. It is also
possible that experimentally the two symmetric nitro
stretch modes are degenerate, with both contributing
to the observed peak.
4. Conclusions
The use of the B3LYP functional with the triple
zeta, 6-311 þ G** basis set gives accurate frequen-
cies and IR intensities for nitrobenzene and accounts
for the isotopic shifts observed for the isotopomers,
without the need to rescale the force constants. The
present analysis improves the previous analysis by
Shlyapochinkov et al. [15], accounting for all the
observed fundamentals, plus Fermi resonance, com-
binations and overtones below 1650 cm21
, with the
new data from nb-13
C6 helping in the assignment of
the spectra.
References
[1] L. Quaroni, W.E. Smith, J. Raman. Spectrosc. 30 (1999)
537.
[2] L. Quaroni, J. Reglinski, R. Wolf, W.E. Smith, Biochim.
Biophys. Acta 1296 (1996) 5.
[3] L. Quaroni, W.E. Smith, Biospectroscopy 5 (1999) S71.
[4] V.G. Osipov, V.A. Shlyapochnikov, E.F. Ponizovtsev, Zh.
Prikl. Spektosk. 8 (1968) 1003.
[5] J.H.S. Green, W. Kynaston, A.S. Lindsay, Spectrochim. Acta
17 (1961) 933.
[6] C.V. Stephenson, W.C. Coburn, W.S. Wilcox, Spectrochim.
Acta 17 (1961) 486.
[7] S. Daehne, H. Stanko, Spectrochim. Acta 18 (1962) 561.
[8] S. Pinchas, D. Samuel, B.L. Silver, Spectrochim. Acta 20
(1964) 179.
[9] W.R. McWhinnie, R.C. Poller, Spectrochim. Acta 22 (1966)
501.
[10] V.C. Farmer, Spectrochim. Acta, Part A 23 (1967) 728.
[11] G. Varsanyi, S. Holly, L. Imre, Spectrochim. Acta, Part A 23
(1967) 1205.
[12] J.H.S. Green, D.J. Harrison, Spectrochim. Acta, Part A 26
(1970) 1925.
[13] A. Kuewae, K. Machida, Spectrochim. Acta, Part A 35 (1979)
27.
[14] J.D. Laposa, Spectrochim. Acta, Part A 35 (1979) 65.
[15] V.A. Shlyapochnikov, L.S. Khaikin, O.E. Grikina, C.W. Bock,
L.V. Vilkov, J. Mol. Struct. 326 (1994) 1.
[16] A. Kova´cs, V. Izvekov, G. Keresztury, G. Pongor, Chem.
Phys. 238 (1998) 231.
[17] J. Clarkson, W.E. Smith, D.N. Batchelder, D.A. Smith, A.M.
Coats, J. Mol. Struct. 648 (2003) 203.
[18] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A.
Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery,
Jr., R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam,
A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi,
V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.
Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,
Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck,
K. Raghavachari, J.B. Foresman, J. Cioslowski, J. V. Ortiz,
A.G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,
I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith,
M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez,
M. Challacombe, P.M.W. Gill, B.G. Johnson, W. Chen, M.W.
Wong, J.L. Andres, M. Head-Gordon, E.S. Replogle, J.A.
Pople, GAUSSIAN 98 (Revision A.9), Gaussian, Inc., Pittsburgh
PA, 1998.
[19] A. Becke, J. Chem. Phys. 98 (1993) 5648.
[20] C. Lee, W. Wang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[21] W.J. Hehre, R. Ditchfield, J.A. Pople, J. Chem. Phys. 56
(1972) 2257.
[22] P.C. Hariharan, A.J. Pople, Theor. Chim. Acta 28 (1973) 213.
[23] M.S. Gordon, Chem. Phys. Lett. 76 (1980) 163.
[24] A.D. McLean, G.S. Chandler, J. Chem. Phys. 72 (1980)
5639.
[25] R. Krishnan, J.S. Binkley, R. Seeger, A.J. Pople, J. Chem.
Phys. 72 (1980) 650.
[26] E.B. Wilson, Phys. Rev. 45 (1934) 706.
[27] G. Varsanyi, Vibrational Spectra of Benzene Derivatives,
Academic Press, New York, 1969.
J. Clarkson, W. Ewen Smith / Journal of Molecular Structure 655 (2003) 413–422422](https://image.slidesharecdn.com/adftanalysisofthevibrationalspectraofnitrobenzene-150806102345-lva1-app6892/75/A-dft-analysis-of-the-vibrational-spectra-of-nitrobenzene-10-2048.jpg)

The document analyzes the vibrational spectra of nitrobenzene and its isotopomers using density functional theory calculations. It finds that a B3LYP/6-311+G** calculation can accurately model the vibrational spectra and isotopic shifts without scaling force constants. This represents an improvement over previous methods that required such scaling. The calculation reproduces the experimental Raman and infrared spectra and allows reassignment of some vibrational modes.
























































