Download to read offline
![A theoretical study of the structure and vibrations
of 2,4,6-trinitrotolune
John Clarksona,*, W. Ewen Smitha
, David N. Batchelderb
, D. Alastair Smithb
,
Alison M. Coatsc
a
Department of Pure and Applied Chemistry, Strathclyde University, Thomas Graham Building, 295 Cathedral Street,
Glasgow G1 1XL, Scotland, UK
b
Department of Physics and Astronomy, Leeds University, Leeds LS2 9JT, UK
c
Department of Chemistry, University of Aberdeen, Meston Walk, Aberdeen AB24 3UE, Scotland, UK
Received 21 October 2002; revised 13 December 2002; accepted 13 December 2002
Abstract
Theoretical calculations of the structure, internal rotations and vibrations of 2,4,6-trinitrotolune, TNT, in the gas phase were
performed at the B3LYP/6-31G* and B3LYP/6-311 þ G** levels of theory. Two genuine energy minimum structures were
found. In both structures the 4-nitro group is planar to the phenyl ring, while the 2,6-nitro groups are slightly out of plane with
the phenyl ring due to steric interaction with the methyl group. The two structures are related by internal rotations of the methyl
and 2, or 6-nitro group. The lowest energy route for interconversion between them is a concerted motion of the methyl group
and 2 or 6 nitro group in a ‘cog wheel’ type of mechanism. The geometry of the low energy structure A is closest to that
observed in the crystal structures of TNT, where all three nitro groups are out of plane with the phenyl ring. FTIR and Raman
spectra of solid TNT and 13
C, 15
N enriched TNT are presented and assigned with the help of the B3LYP/6-311 þ G**
calculations on A. The lower level B3LYP/6-31G* calculation fails to predict the correct vibrational coupling between the nitro
and phenyl groups. The B3LYP/6-311 þ G** calculation gives a good prediction of the nitro vibrations and the isotopic shifts
observed for TNT isotopomers.
q 2003 Elsevier Science B.V. All rights reserved.
Keywords: Trinitrotoluene; Density functional theory; Vibrational analysis
1. Introduction
The structure of aromatic nitro compounds has
been of great interest due to the explosive nature of
these compounds [1]. 2,4,6-Trinitrotoluene, TNT, is
one of the best known and widely used explosive
materials and has been extensively studied [2–20].
To understand the explosive nature of TNT, the
molecular geometry needs to be examined in detail
as this can inform on possible mechanisms and
routes of thermal decomposition [21,22]. Density
functional theory (DFT) methods has recently been
successfully applied to investigate the structure and
vibrations of two other, well known explosive
0022-2860/03/$ - see front matter q 2003 Elsevier Science B.V. All rights reserved.
doi:10.1016/S0022-2860(03)00024-3
Journal of Molecular Structure 648 (2003) 203–214
www.elsevier.com/locate/molstruc
* Corresponding author. Tel.: þ44-141-552-4400; fax: 44-141-
552-0876.
E-mail address: john.clarkson@strath.ac.uk (J. Clarkson).](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-1-2048.jpg)
![compounds, 1,3,5-trinitro-s-triazine (RDX) [23] and
pentaerythritol tetranitrate (PETN) [24]. Here we
present a similar DFT investigation into the
structure and vibrational properties of TNT. We
also present detailed Raman and infrared data for
solid TNT and its 13
C, 15
N enriched isotopomer
and assign this data with the aid of DFT
calculations.
Fig. 1. The theoretical molecular structure of TNT. Molecule A has near ideal Cs symmetry and has lower energy than molecule B.
Table 1
Predicted bond distances for theoretical TNT molecules A and B, together with TNT crystal structure parameters
Distance TNT monoclinica
TNT
orthorhombica
TNT complex
with pyreneb
B3LYP/6-31G* B3LYP/6-
311 þ G**
A B A B A B A B A B
C1–C2 1.399 1.396 1.401 1.409 1.388 1.383 1.411 1.413 1.406 1.407
C2–C3 1.386 1.381 1.367 1.378 1.395 1.367 1.391 1.391 1.387 1.387
C3–C4 1.372 1.372 1.386 1.379 1.346 1.352 1.386 1.386 1.385 1.385
C4–C5 1.375 1.380 1.366 1.358 1.344 1.360 1.386 1.386 1.385 1.384
C5–C6 1.382 1.384 1.394 1.403 1.362 1.366 1.391 1.391 1.387 1.388
C1–C6 1.384 1.394 1.383 1.385 1.399 1.385 1.411 1.410 1.406 1.405
C1–C7 1.510 1.502 1.500 1.524 1.507 1.532 1.509 1.509 1.506 1.506
C7–H8 0.989 0.983 1.002 0.960 0.781 0.879 1.089 1.093 1.088 1.091
C7–H9 0.898 0.949 0.918 0.964 1.101 1.305 1.094 1.086 1.092 1.084
C7–H10 0.985 0.940 0.914 1.006 0.936 1.080 1.090 1.094 1.088 1.092
C2–N11 1.474 1.478 1.471 1.475 1.477 1.462 1.481 1.482 1.485 1.485
N11–O12 1.228 1.219 1.219 1.198 1.232 1.197 1.228 1.228 1.222 1.222
N11–O13 1.215 1.214 1.226 1.231 1.187 1.204 1.228 1.228 1.221 1.221
C3–H14 0.978 1.002 0.933 1.062 0.764 1.079 1.082 1.082 1.081 1.081
C4–N15 1.470 1.462 1.465 1.463 1.477 1.492 1.475 1.475 1.482 1.482
N15–O16 1.216 1.219 1.240 1.199 1.211 1.199 1.228 1.228 1.221 1.221
N15–O17 1.212 1.227 1.210 1.222 1.213 1.244 1.228 1.228 1.221 1.221
C5–H18 0.967 0.977 1.029 1.103 0.902 1.078 1.082 1.082 1.081 1.081
C6–N19 1.483 1.475 1.475 1.466 1.486 1.483 1.481 1.483 1.485 1.488
N19–O20 1.215 1.218 1.240 1.222 1.222 1.219 1.228 1.228 1.222 1.221
N19–O21 1.208 1.214 1.171 1.214 1.214 1.181 1.228 1.227 1.221 1.221
a
Data from Ref. [15].
b
Data from Ref. [16]. Two molecules in the unit cell.
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214204](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-2-2048.jpg)
![Recent structural X-ray studies on TNT have
shown the existence of two crystallographic forms,
monoclinic and orthorhombic [14–17]. The nitro
groups of TNT all adopt a non-planar configuration
due to crystal packing forces, extensive inter and
intramolecular van der Waals interactions and also
steric effects of the methyl group. The structure and
vibrations of TNT had been previously studied in a
series of papers by Carper and Stewart using
semiempirical theories aided by infrared data from
a number of TNT isotopomers [8–11]. These
calculations that predict non-planar conformations
for all the nitro groups, however, do not predict
accurate vibrational frequencies. A short communi-
cation using the Wilson GF matrix method with the
Urey–Bradley force field on the crystal structure
of TNT does report accurate nitro group
vibrations [12]. Recent ab initio Hartree–Fock,
HF, calculations appear to account for the steric
effects of the methyl group, with the 2 and 6 nitro
groups predicted to be non-planar and the 4-nitro
group planar to the phenyl ring [6,7]. The HF
calculation for TNT, however, does not predict
accurate nitro vibrations [7], an error in common
with HF calculations of other aromatic nitro
compounds [25,26]. DFT calculations of a range
of aromatic nitro compounds show that these
methods can predict accurate nitro vibrations
[26–29].
The DFT calculations for RDX and PETN both
predict structures essentially identical in geometry
to those observed in the solid state [23,24]. This
good prediction of molecular structure allows
for good prediction of the normal modes and
Table 2
Predicted angles for theoretical TNT molecules A and B, together with TNT crystal structure parameters
Angle TNT monoclinic TNT
orthorhombic
TNT complex
with pyrene
B3LYP/6-31G* B3LYP/6-
311 þ G**
A B A B A B A B A B
C2–C1–C6 113.6 113.7 113.8 114.0 113.4 114.4 114.2 114.1 113.9 113.8
C1–C2–C3 124.6 125.1 124.5 124.8 122.6 124.2 124.0 124.0 124.3 124.3
C2–C3–C4 116.9 116.6 117.6 116.2 119.2 117.6 118.3 118.3 118.0 117.9
C3–C4–C5 122.8 122.8 122.1 124.0 121.5 122.1 121.2 121.3 121.5 121.5
C4–C5–C6 116.7 117.1 116.9 116.6 118.5 118.3 118.5 118.3 118.0 118.0
C5–C6–C1 125.3 124.4 124.9 124.3 124.7 123.3 123.7 124.0 124.3 124.3
C1–C7–H8 111.3 111.8 99.6 104.3 112.1 110.6 111.1 110.9 110.9 111.3
C1–C7–H9 110.2 111.2 125.3 98.9 110.5 104.6 110.5 111.4 110.4 111.7
C1–C7–H10 115.2 110.2 125.4 112.1 102.1 111.6 111.2 109.7 111.1 109.6
C1–C2–N11 119.9 118.8 120.0 117.8 121.2 121.8 121.2 121.3 120.4 120.4
C3–C2–N11 115.4 116.1 115.5 117.4 116.2 114.0 114.8 114.8 115.3 115.2
C2–N11–O12 118.0 117.1 118.4 120.3 117.0 119.4 117.7 118.1 117.5 117.4
C2–N11–O13 117.3 117.6 116.5 119.4 119.0 119.0 117.0 116.8 116.9 116.8
O12–N11–O13 124.7 125.2 125.1 120.5 123.9 121.5 125.2 125.1 125.6 125.8
C2–C3–H14 120.5 125.5 124.7 117.4 124.9 121.1 120.9 120.9 121.3 121.0
C3–C4–N15 118.3 118.2 118.9 117.2 119.6 118.5 119.5 119.3 119.3 119.2
C5–C4–N15 118.9 118.9 118.8 118.7 118.9 119.4 119.3 119.3 119.2 119.2
C4–N15–O16 117.5 118.1 117.4 118.7 117.7 116.9 117.1 117.2 117.1 117.2
C4–N15–O17 117.7 117.3 117.5 117.1 117.6 115.9 117.3 117.2 117.1 117.1
O16–N15–O17 124.7 124.6 125.1 124.1 124.7 127.3 125.4 125.6 125.8 125.8
C4–C5–H18 115.9 122.3 112.2 127.1 125.0 121.2 120.8 120.8 121.0 120.6
C5–C6–N19 115.7 115.3 114.6 115.4 115.2 115.9 115.0 114.1 115.4 114.6
C6–N19–O20 117.8 117.9 115.1 118.4 117.9 118.5 117.7 118.1 117.3 117.6
C6–N19–O21 117.1 117.5 116.3 118.0 117.3 118.2 117.0 116.5 117.1 116.6
O20–N19–O21 125.1 124.6 128.3 123.5 124.7 123.1 125.3 125.3 125.6 125.7
C1–C6–N19 119.1 120.3 120.5 120.4 120.1 120.9 121.3 121.9 120.4 121.1
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214 205](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-3-2048.jpg)
![assignment of the vibrational spectra. The solid
state structure of TNT reveals a molecular structure
subject to intramolecular forces and in particular all
three nitro groups are non-planar to the phenyl ring
[14–17]. The present DFT calculations reveals that
a good prediction of the normal modes of TNT
can, however, still be obtained despite differences
between the solid state structure and the theoretical
gas phase structure.
2. Methods
TNT was supplied as a gift from Professor John
N. Sherwood (Strathclyde University) as single
crystals, as used in previous studies [18,20]. 13
C
(99%), 15
N (98%) enriched TNT was obtained from
Cambridge Isotope Laboratories, Inc. A crystal of
TNT was crushed to a powder prior to obtaining a
Raman spectrum. A thin plate of TNT was similarly
prepared for FTIR analysis.
Raman spectra were obtained using a Renishaw
micro-Raman system 1000 spectrometer
with 785 nm excitation. FTIR spectra were
obtained using a Nicolet Nexus spectrometer
with a Spectra-Tech Continuum microscope
attachment.
Geometry optimization and frequency analysis
of TNT was performed using GAUSSIAN98 [30]
with the B3LYP hybrid density functional [31,32]
using the 6-31G* [33–35] and 6-311 þ G** [36,
37] basis sets. Two genuine energy minimum
structures were found, A and B, Fig. 1, as verified
by vibrational frequency calculations; no negative
frequencies were found.
3. Results and discussion
3.1. Structure and internal rotations of TNT
The two stable molecular structures of TNT,
Fig. 1, are related by internal rotations of the 2 and
6-nitro groups and the methyl group. The calculated
bond lengths and angles are compared to those
observed in the crystal structures of TNT [15] in
Tables 1 and 2. The geometry of the solid state
structures are subject to intramolecular forces, such
as van der Waals interactions and crystal packing
forces with the most notable difference between
these and the calculated structures are the dihedral
angles of the nitro groups Table 3. A comparison of
the calculated structures of TNT with the structures
observed experimentally [15] shows that structure A
has a geometry closest to those observed exper-
imentally. The TNT molecules co-crystallized with
pyrene [16] display a structure that is even closer to
A than the molecules in the monoclinic and
orthorhombic crystals of pure TNT [15]. In all the
solid state structures of TNT, the three nitro groups
are non-planar to the phenyl ring, however, the
4-nitro group is not twisted far from planarity and
the 2 and 6-nitro groups point to the same face of
the phenyl ring in a manner close to that modeled in
Table 3
Selected dihedral angles for theoretical TNT molecules A and B, together with TNT crystal structure parameters
Dihedral angle TNT monoclinic TNT
orthorhombic
TNT complex with
pyrene
B3LYP/6-31G* B3LYP/6-
311 þ G**
A B A B A B A B A B
O12–N11–C2–C1 47.2 59.5 42.1 53.1 29.4 39.3 33.4 31.8 41.1 41.0
O16–N15–C4–C3 22.0 232.2 35.3 223.7 20.8 0.2 0.1 1.1 0.2 0.8
O20–N19–C6–C1 251.2 240.9 257.5 246.4 243.4 227.2 233.5 45.4 241.1 52.2
O12–N11–N19–O20 25.8 20.7 218.1 7.9 212.5 11.8 20.1 69.7 0.1 83.7
H9–C7–C1–C6 97.6 86.4 117.7 76.7 92.28 87.4 91.8 214.2 91.8 216.1
Table 4
Calculated energies for theoretical molecules TNT A and B in
Hartrees
Molecule B3LYP/6-31G* B3LYP/6-311 þ G**
A 2885.045504504 2885.303387940
B 2885.044182860 2885.302350910
DE 0.829 kcal Mol21
0.650 kcal Mol21
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214206](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-4-2048.jpg)
![A. The TNT molecules co-crystallized with pyrene
also display less intermolecular-van der Waals
interactions than pure TNT. No vapor phase
structural data is available for TNT, which would
shed further light on the true molecular geometry of
isolated TNT molecules. In dilute solution TNT may
adopt structure A, though the barriers to internal
rotations to structure B, are small, see below.
Structure B is identical to that found by Chen in his
HF/6-31G* calculation [6] and structure A is identical
to that reported by Janni et al. in a similar HF/6-31G*
calculation [7]. These previous studies did not explore
other possible structures of TNT, though Janni et al.
[7] did note the difference in the 2,6-nitro group
dihedral angles they calculated compared to those
calculated by Chen [6].
Structure A is the more stable geometry, though
the difference in energy is small (0.650 kcal mol21
at B3LYP/6-311 þ G**, Table 4). Structure A
displays near ideal Cs symmetry with one of the
methyl hydrogen atoms perpendicular to the phenyl
ring in the plane of sh: The 2,6-nitro groups of A
are non-planar and rotated into the same face of the
phenyl ring, maximizing the number of van der
Waals interactions to the methyl group. These nitro
groups, examined from the side of the phenyl ring,
eclipse each other, with the B3LYP/6-311 þ G**
dihedral angle O12–N11–N19–20 calculated to be
near zero, 20.18 Table 3. The 4-nitro groups of A
and B are calculated to be planar with the phenyl
ring at all levels of theory, thus maximizing the
conjugation of the nitro group to the phenyl ring.
The steric hindrance between the methyl and
2,6-nitro groups prevents the 2,6-nitro groups
from adopting a planar geometry. It is this steric
hindrance that is the reason TNT can adopt the two
structures shown in Fig. 1. Structure B is less
stable than A due to the 6-nitro group being more
twisted out of the plane of the phenyl ring,
reducing the conjugation to the phenyl ring. The
2-nitro group in structure B, however, adopts a
geometry near identical to that of structure A.
Structure B also displays one less intra-molecular
van der Waals interaction with the methyl group
Table 5. The extra van der Waals interaction
together with increased planarity of the 6-nitro
group contributes to the increased stability of
structure A over B.
Conversion of A to B involves internal rotations
of the methyl group and the 2,6-nitro groups, with
the barrier to such rotations expected to be low
Fig. 2. Relaxed potential energy scans of TNT along the coordinate
corresponding to a 2 or 6-nitro group rotation of 1808. Points A and
B/B0
correspond to the structures of molecules A and B in Fig. 1.
Table 5
Selected van der Waals distances for theoretical TNT molecules A and B, together with TNT crystal structure parameters
van der Waal distance TNT monoclinic TNT
orthorhombic
TNT complex
with pyrene
B3LYP/6-31G* B3LYP/6-
311 þ G**
A B A B A B A B A B
O12–H10 2.618 3.011 2.530 2.571 2.332 2.454 2.343 2.514 2.424 2.577
O12–H9 2.798 2.689 2.719 2.961 2.813 2.937 2.821 2.852
O12–H8 2.499 2.582
O13–H14 2.574 2.830 2.533 2.701 2.543 2.431 2.422 2.399 2.513 2.506
O16–H14 2.490 2.481 2.563 2.538 2.385 2.382 2.420 2.422 2.423 2.426
O17–H18 2.396 2.571 2.341 2.548 2.471 2.391 2.420 2.416 2.423 2.425
O21–H18 2.722 2.520 2.699 2.528 2.461 2.387 2.223 2.499 2.513 2.419
O20–H8 2.273 2.525 2.653 2.578 2.603 2.220 2.342 2.424
O20–H9 2.942 2.789 3.426 2.684 2.848 2.787 2.821 2.170 2.852 2.240
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214 207](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-5-2048.jpg)
![[38–40]. Relaxed potential energy scans, with full
geometric HF/6-31G* optimization except for a
fixed constraint along the coordinate corresponding
to a 2 or 6-nitro group rotation is shown on Fig. 2.
Structure A has a nitro angle at 358. Both scans
reveal two barriers between A and B/B0
at 90 and
1708. The large barrier at 908 is due to the lack of
conjugation between the nitro and phenyl groups; A
to B0
. The 1708 feature is at the apex of a barrier
of a cogwheel type of motion involving a concerted
motion between the nitro and methyl group; A to
B. This cogwheel motion is similar to that modeled
for the rotation of the adjacent nitro groups in 1,2-
dintrobenzene [38]. The profile of the scan between
120 and 1608 depends on which direction the nitro
group is rotated. Steps to the positive side result in
a minimum at 1358, B0
, and negative steps result in
a minimum at 1458, B. These minima represent
identical structures, B, and are related by a methyl
rotation of 608, together with a change in dihedral
angle of the other nitro group adjacent to the
methyl group. The curves beyond the 135/145
minima that suddenly drop to trace the smooth
barriers between A and B/B0
correspond to saddle
edges on the potential energy surface. This
potential energy surface can be examined qualitat-
ively by taking single point energy values, (HF/
STO-3G), between a range of fixed values for the
rotation angles of the 2 (or 6) nitro and methyl
groups, while all other geometric parameters are
frozen at the HF/6-31G* level. The resulting
energy landscape, Fig. 3 reveals three minima,
corresponding to structures A and B. Minimum B0
is essentially identical to minimum B and has a
slightly higher energy due to the constraints
imposed on the molecular geometry. The saddle
edges seen in Fig. 2 must be along the two high
energy features, 1 and 2, which are due to the
steric interaction between the methyl and
nitro group, 1, and the 908 rotation of the nitro
group, 2.
The predicted barrier to the nitro rotation at 908
(point H1, Fig. 2), is 2.54 kcal mol21
in height
from A, similar to that calculated for nitrobenzene
and other 2-substituted nitrobenzene molecules
[38–40]. The small barrier (point H2, Fig. 2), is
1.59 kcal mol21
in height from A. The heights of
the energy barriers from B to A are
1.29 kcal mol21
via H1 and 0.34 kcal mol21
via
H2. The small heights to these barriers suggest that
TNT molecules, in the gas phase, at room
temperature are readily able to inter-convert
between structures A and B.
Fig. 4. Infrared and Raman spectra of TNT (solid line) and 13
C, 15
N-
TNT (dotted line).
Fig. 3. A constrained potential energy surface scan of TNT along the
coordinates corresponding to a methyl group rotation of 1208 and a
2 or 6-nitro group rotation of 1808. Points A and B/B0
correspond to
the structures of molecules A and B in Fig. 1. 1 reflects lack of
conjugation between the nitro and phenyl group and 2 reflects steric
interaction between the methyl and nitro group.
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214208](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-6-2048.jpg)
![3.2. Vibrational analysis
There is extensive FTIR data in the literature for
TNT and a variety of it is isotopomers [4-(15
NO2)-
TNT, (15
NO2)3-TNT, (15
NO2)3-TNT-d5, TNT-a-d3
and TNT-d5] [8–11]. To this data set we now add
Raman and FTIR data for solid TNT and solid 13
C,
15
N-TNT Fig. 4. This is a particularly rich database
for such a large molecule as TNT and a comparison
with predicted theoretical isotopic shifts allows the
modeled normal modes to be assessed for accuracy.
Vibrational frequencies were calculated for all
levels of theory for structures A and B. Table 6
displays the predicted symmetric and asymmetric
nitro vibrational frequencies for each level of theory.
All theoretical frequencies reported here are listed as
calculated, as no scale factor is available for the
B3LYP functional with the 6-311 þ G** basis set.
A scale factor of 0.9613 is recommended for B3LYP/
6-31G* [41], however, as discussed below this level
of theory does not give accurate modes of vibration
for TNT and the frequencies listed on Table 6 are not
scaled. There is very little difference between the
asymmetric and symmetric nitro frequencies for A
and B, though the mode character is different for a
few. Table 7 shows the assignment of the asymmetric
and symmetric nitro vibrations for TNT and its
isotopomers based on the normal mode analysis of
A, which is found to be similar to the solid state
structure. The calculation gives reasonable prediction
of the normal mode frequencies and the observed
isotopic shifts. This is despite differences between the
solid state structure and the theoretical structure,
discussed above.
Table 8 lists the B3LYP/6-311 þ G** predicted
frequencies for molecules A and B together with
Table 6
Predicted asymmetric and symmetric nitro group vibrational frequencies at various levels of theory
B3LYP/6-31G* TNT A B3LYP/6-31G* TNT B B3LYP/6-311 þ G** TNT A B3LYP/6-311 þ G** TNT B
Asymmetric NO2 1649 A00
1651 1603 A00
1606
1635 A0
1636 1603 A0
1602
1616 A00
1616 1589 A00
1590
Symmetric NO2 1407 A0
1410 1389 A0
1393
1397 A00
1399 1380 A00
1381
1394 A0
1394 1370 A0
1370
Table 7
A comparison of predicted (model A at the B3LYP/6-311 þ G** level of theory) and observed nitro group vibrational frequencies for TNT and
it is various isotopomers
Model A TNTa
4-(15
NO2)-TNT (15
NO2)3-TNT (15
NO2)3-TNT-d5
13
C, 15
N-TNTa
TNT-a-d3 TNT-d5
Asymmetric
NO2
a n50 1603 (1550) n50 1602 n49 1567 (1509) n49 1566 (1507) n50 1567 (1514) n50 1602 n50 1602
b n49 1603 (1545) n49 1601 (1540) n50 1573 n50 1567 n49 1561 (1505) n49 1600 (1539) n49 1596
(1541)
c n48 1589 (1533) n48 1564 (1507) n48 1564 n48 1561 n48 1543 (1497) n48 1587 n48 1581
(1522)
Symmetric
NO2
a n42 1389 (1375) n42 1387 (1375) n42 1366 n45 1366 n42 1365 (1346) n42 1390 n46 1389
b n41 1380 (1365) n41 1380 (1352) n41 1358 n44 1357 n41 1356 (1362) n41 1380 n44 1380
c n40 1370 (1359) n40 1349 (1331) n40 1347 (1329) n43 1347 (1323) n40 1347 (1329) n40 1370 (1354) n43 1369
(1359)
a: 2,4,6-nitro stretching. b: 2,6,-nitro stretching. c: 4-nitro stretching.
a
From this study FTIR; other data from Refs. [8–10].
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214 209](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-7-2048.jpg)
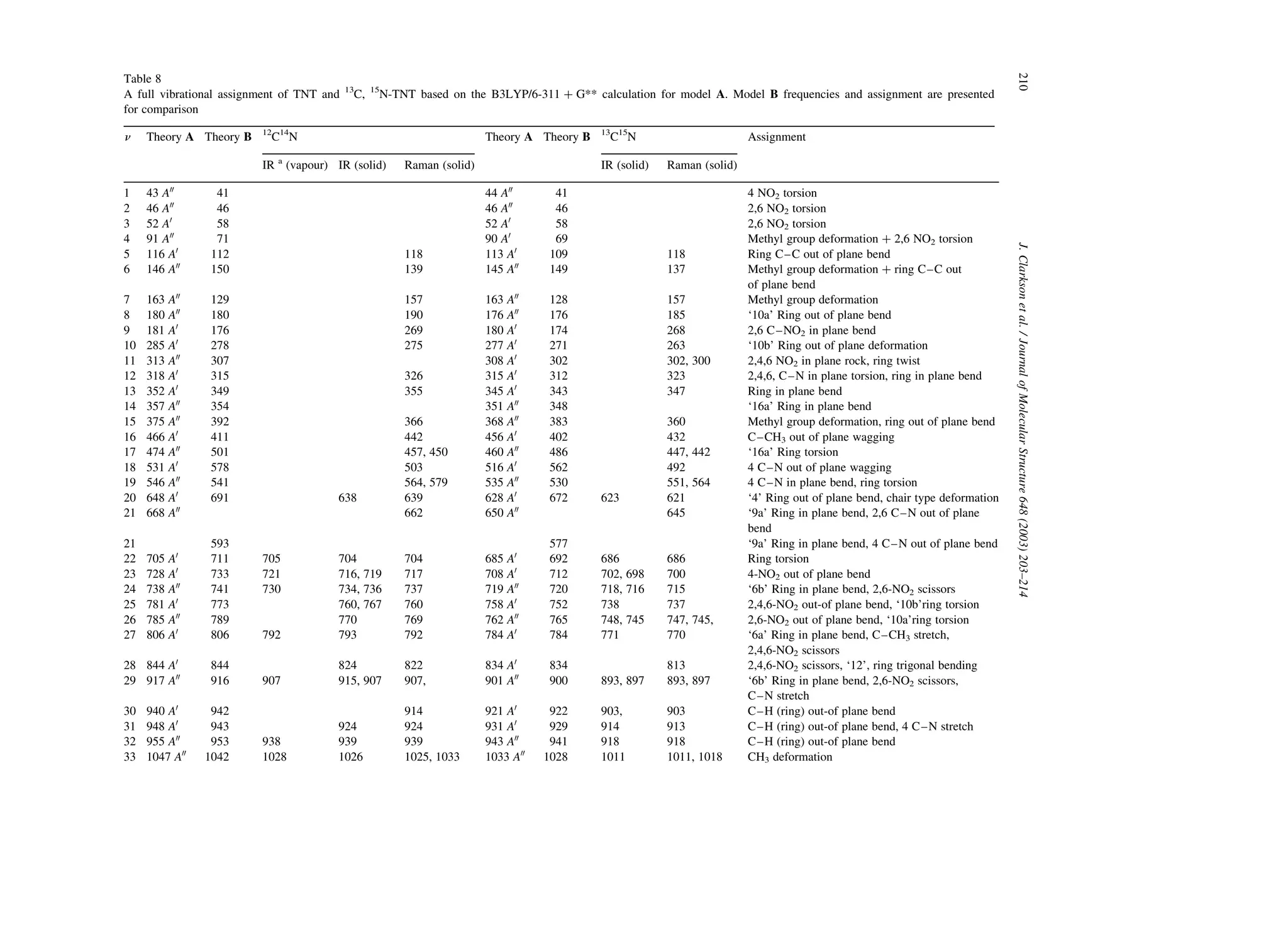
![34 1049 A0
1056 1057 1033 A0
1041 CH3 deformation
35 1097 A0
1097 1067 1086 1090, 1086 1074 A0
1075 1065 1066 C–H (ring) in plane bend
36 1179 A0
1179 1075 1171 1170 1141 A0
1141 1140, 1137 1140, 1138 ‘12/1’ C–C (ring) in plane trigonal
bend, 2,4,6 C–N & C–CH3 stretch
37 1219 A0
1218 1217 1208 1210, 1213 1180 A0
1180 1174 1174 ‘1’, Ring breathing
38 1219 A00
1222 1230 1204 A00
1206 1190 C–H (ring) in plane bend
39 1343 A00
1340 1305 1292, 1305 1294 A00
1291 1264 1305, 1264 ‘14’ Ring stretching (Kekule)
40 1370 A0
1370 1352 1350 1359 1347 A0
1347 1322 1329 4-NO2 Symmetric, C–N stretch
41 1380 A00
1365 1356 A00
1362 2,6-NO2 Symmetric, C–N stretch
41 1381 1358 2,4,6-NO2 Symmetric, C–N stretch
42 1389 A0
1375 1365 A0
1346 2,4,6-NO2 Symmetric, C–N stretch
42 1393 1367 2,4,6-NO2 Symmetric, C–N stretch
43 1419 A0
1416 1381 1381 1409 A00
1409 1372 CH3 umbrella deformation
44 1421 A00
1423 1420 1406 1403 1380 A0
1380 1367 1375 ‘19b’ Ring stretching
45 1475 A0
1471 1437 1437 1438 1435 A0
1434 1419 1418, 1428 ‘19a’,Ring stretching, CH3 deformation
46 1476 A00
1482 1446 1468 A00
1470 1427 1440 CH3 deformation
47 1498 A0
1507 1457 1466 1466 1491 A0
1502 CH3 deformation
48 1589 A00
1590 1508 1533 1534 1543 A00
1543 1496 1497 4-NO2 asymmetric stretch, ‘8b’ ring stretching
49 1603 A0
1539 1545 1545 1561 A0
1505 1505 2,6-NO2 asymmetric stretch, ‘8a’ ring stretching
49 1602 1562 2-NO2 asymmetric stretch, ‘8a’ ring stretching
50 1603 A00
1551 1551 1567 A00
1513 1514 2,4,6-NO2 asymmetric stretching
50 1606 1569 6-NO2 asymmetric stretching
51 1646 A00
1645 1602 1602 1595 A00
1593 1547 1548 ‘8b’ Ring stretching, 4-NO2
asymmetric stretching
52 1651 A0
1617 1617 1618 1596 A0
1561 1561 ‘8a’ Ring stretching, 2,6-NO2
asymmetric stretching
52 1650 1596 ‘8a’ Ring stretching, 2,4,6-NO2
asymmetric stretching
53 3065 A0
3063 2882 3061 A0
3059 C–H (methyl) stretching
54 3128 A0
3113 2912 3118 A0
3102 C–H (methyl) stretching
55 3155 A00
3187 2955 3143 A00
3176 C–H (methyl) stretching
56 3233 A0
3233 3056 3223 A0
3222 C–H (ring) stretching
57 3233A00
3235 3085, 3096 3222 A00
3224 C–H (ring) stretching
a
Data from Ref. [8]
J.Clarksonetal./JournalofMolecularStructure648(2003)203–214211](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-9-2048.jpg)
![the experimental frequencies for TNT and 13
C,
15
N-TNT and an assignment based on visual inspec-
tion of the normal modes. The phenyl ring vibrations
are labeled according to the Wilson notation [42,43].
The experimental data is assigned with the help of A,
as this has the closest geometry to that found
experimentally. The predicted modes of B are
presented for a comparison to A and to allow an
assessment of the affect of the change in molecular
structure. Many of the normal modes of A and B are
found to be very similar in both frequency and
character. The exceptions to this are some modes
involving the methyl and 2,6-nitro groups, especially
the out of plane modes. n21 in particular shows large
difference in frequency due to the different nitro group
contributions to the normal mode. Molecule A shows
contributions from the 2,6 nitro groups for n21,
whereas molecule B shows contributions form the 4
nitro group; the character of the phenyl ring
contribution is essentially identical for both mol-
ecules. The 2,6-nitro groups of A and B correspond to
N11 and N19, respectively, Fig. 2. The loss of
symmetry between the 2 and 6 nitro groups upon
going from molecule A to B causes redistribution in
the coupling of these groups in the normal modes.
This is demonstrated most clearly in the asymmetric
and symmetric nitro vibrations.
The lower level B3LYP/6-31G* calculation pre-
dict the highest non C–H stretching frequency to be
dominated by nitro asymmetric stretching, coupled to
Fig. 5. The predicted B3LYP/6-31G*, B3LYP/6-311 þ G** and
experimental IR data for TNT. 1: Phenyl ring modes 8a and 8b. 2:
Asymmetric nitro modes. 3: Symmetric nitro modes.
Fig. 6. The B3LYP/6-311 þ G** eigenvectors for the highest non
C–H stretching modes and the three asymmetric nitro vibrational
modes of TNT, n52–n48; the three symmetric nitro vibrational
modes, n42–n40 and n28–n29. The differing lengths of the arrow
show the relative differences in amplitude of the vibrating atom.
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214212](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-10-2048.jpg)
![the phenyl ring 8a/8b modes and have a large IR
intensity, Fig. 5. The B3LYP/6-311 þ G** calcu-
lation correctly predicts the two highest non C–H
stretching modes to be dominated by phenyl ring
vibrations and the frequencies at 1616 and 1602,
Fig. 5, are assigned to the phenyl ring stretching
modes 8a (n52) and 8b (n51), respectively. These
modes also have a small contribution from the
asymmetric nitro stretches, most notable n51 Fig. 6.
Three nitro asymmetric vibrations are predicted,
n50–48n Fig. 6. n50 is dominated by the nitro groups
with little contribution from the phenyl ring, whereas
n49 and n48, are coupled to the phenyl ring modes 8a
and 8b, respectively. Visual inspection of the n49 and
n48 modes reveal that they are similar to n50 and n51,
Fig. 6, with only the degree of nitro and phenyl ring
8a/b contribution being different. There are notable
differences in the normal modes character of the n50
and n49 between molecules A and B, due to molecular
symmetry and vibrational coupling differences
between nitro and phenyl groups. The three predicted
symmetric nitro group vibrations, n42–n40, reveal a
similar pattern in nitro group participation to that
observed for the asymmetric vibrations. That is,
molecule A shows a pattern of nitro group partici-
pation of (2,4,6), (2,6), (4), while B shows a pattern of
(6), (2), (4), nitro dominated modes. The different
geometry of the 2,6 nitro groups between A and B
does not greatly affect the frequencies of the
asymmetric and symmetric nitro modes. n48 and
n40 both involve strong contributions from the 4-nitro
group and the FTIR data from 4-(15
NO2)-TNT is
particularly relevant in assigning these modes. The
TNT bands at 1533 and 1359 cm21
are assigned to
n48 and n40, respectively, on the basis of the observed
isotopic shifts, which are reasonably well modeled by
the B3LYP/6-311 þ G** calculation of A Table 7.
The two intense Raman bands at 822 and
792 cm21
, assigned to n28 and n29, respectively,
both have contributions from NO2 scissors, especially
the 822 cm21
band, where the NO2 scissors is highly
coupled to the phenyl ring trigonal bending mode 12.
The agreement between the predicted and exper-
imental vibrational frequencies is quite good,
especially for those modes not involving the nitro
groups. The new vibrational data we present for 13
C,
15
N-TNT is also assigned and well modeled well by
the B3LYP/6-311 þ G** calculation for model
A Table 8. In a similar fashion to simple [44,45]
and large aliphatic nitro containing molecules [23,24]
the use of the B3LYP hybrid density functional with a
large triple zeta basis set allows the vibrational spectra
of even large aromatic molecules such as TNT to be
assigned with confidence.
4. Conclusions
A theoretical analysis of the structure of TNT
reveals two stable structures related by internal
rotations of the 2 or 6 nitro group and the methyl
group. The geometry of the lower energy structure, A,
is close to that observed in the crystal structure. The
predicted nitro group vibrations are sensitive to the
basis set used, with the B3LYP/6-311 þ G** calcu-
lation for A giving reasonable agreement with
observed frequencies and isotopic shifts, despite
differences between the solid state and the modeled
structure. Many of the predicted normal modes of A
and B are similar, except those dominated by the
methyl and nitro groups, and even then the frequen-
cies are often similar. The fit between experimental
and theoretical frequencies is generally very good
with the highest error being for those modes involving
the nitro and methyl groups due to the difference
between the solid state and theoretical TNT structures.
References
[1] T. Urbanski, Chemistry and Technology of Explosives,
Pergamon Press, New York, 1984.
[2] P.S. Makashir, E.M. Kurian, J. Therm. Anal. Calorim. 55
(1999) 173.
[3] T.B. Brill, K.J. James, J. Phys. Chem. 97 (1993) 8759.
[4] J.C. Oxley, J.L. Smith, H. Ye, R.L. Mckenney, P.R. Bolduc,
J. Phys. Chem. 99 (1995) 9593.
[5] P.S. Makashir, E.M. Kurian, J. Therm. Anal. Calorim. 55
(1999) 173.
[6] P.C. Chen, J. Chin. Chem. Soc. 42 (1995) 755.
[7] J. Janni, B.D. Gilbert, R.W. Field, J.I. Steinfeld, Spectrochim.
Acta A 53 (1997) 1375.
[8] J.J.P. Stewart, S.R. Bosco, W.R. Carper, Spectrochim. Acta
42A (1986) 13.
[9] W.R. Carper, S.R. Bosco, J.J.P. Stewart, Spectrochim. Acta
42A (1986) 461.
[10] W.R. Carper, J.J.P. Stewart, Spectrochim. Acta 43A (1987)
1249.
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214 213](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-11-2048.jpg)
![[11] C.P. Nash, T.E. Nelson, J.J.P. Stewart, W.R. Carper, Spectro-
chim. Acta 45A (1989) 585.
[12] N. Wadhwani, S.G. Wadwani, V.D. Gupta, Def. Sci. J. 44
(1994) 61.
[13] I.R. Lewis, N.W. Daniel, P.R. Griffiths, Appl. Scpectrosc. 51
(1997) 1854.
[14] W.R. Carper, I.P. Davis, M.W. Extine, J. Phys. Chem. 86
(1982) 459.
[15] N.I. Golovina, A.N. Titkov, A.V. Raevskii, L.O. Atovmyan,
J. Solid State Chem. 113 (1994) 229.
[16] J.C. Barnes, W. Golnazarins, Acta Crystallogr. C43 (1987)
549.
[17] P.F. Gerard, A. Hardy, Acta Crystallogr. C44 (1988) 1283.
[18] H.G. Gallagher, J.N. Sherwood, J. Chem. Soc., Faraday Trans.
92 (1996) 2107.
[19] H.G. Gallagher, K.J. Roberts, J.N. Sherwood, L.A. Smith,
J. Mater. Chem. 7 (1997) 229.
[20] R.M. Vrcelj, H.G. Gallagher, J.N. Sherwood, J. Am. Chem.
Soc. 123 (2001) 2291.
[21] J.R. Cox, I.H. Hillier, Chem. Phys. 124 (1988) 39.
[22] T.B. Brill, K.J. James, Chem. Rev. 93 (1993) 2667.
[23] B.M. Rice, C.F. Chabolowski, J. Phys. Chem. A 101 (1997)
8720.
[24] Y.A. Gruzdkov, Y.M. Gupta, J. Phys. Chem. A 105 (2001)
6197.
[25] V.A. Shlyapochnikov, L.S. Khaikin, O.E. Grikina, C.W. Bock,
L.V. Vilkov, J. Mol. Struct. 326 (1994) 1.
[26] P. Urbanowicz, T. Kupka, R. Wrzaliki, K. Pasterny, J. Mol.
Struct. 482–483 (1994) 409.
[27] I.F. Shishkov, L.V. Vilkov, A. Kovacs, I. Hargitta, J. Mol.
Struct. 445 (1998) 259.
[28] V. Kovacs, V. Izveko, G. Keresztury, G. Pongor, Chem. Phys.
238 (1998) 231.
[29] T. Vladimiroff, J. Mol. Struct. 453 (1998) 119.
[30] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A.
Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery
Jr., R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam,
A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi,
V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.
Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,
Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck,
K. Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz,
A.G. Baboul, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,
I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith,
M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez,
M. Challacombe, P.M.W. Gill, B.G. Johnson, W. Chen, M.W.
Wong, J.L. Andres, M. Head-Gordon, E.S. Replogle and J.A.
Pople, GAUSSIAN 98 (Revision A.9), Gaussian Inc., Pittsburgh
PA, 1998.
[31] A. Becke, J. Chem. Phys. 98 (1993) 5648.
[32] C. Lee, W. Wang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[33] W.J. Hehre, R. Ditchfield, J.A. Pople, J. Chem. Phys. 56
(1972) 2257.
[34] P.C. Hariharan, A.J. Pople, Theor. Chim. Acta 28 (1973) 213.
[35] M.S. Gordon, Chem. Phys. Lett. 76 (1980) 163.
[36] A.D. McLean, G.S. Chandler, J. Chem. Phys. 72 (1980) 5639.
[37] R. Krishnan, J.S. Binkley, R. Seeger, A.J. Pople, J. Chem.
Phys. 72 (1980) 650.
[38] J.K. Laerdahl, K. Faegrin, C. Romming, O. Swang, T.
Midtard, K. Schoffel, J. Mol. Struct. 445 (1998) 89.
[39] M. Staikova, I.G. Csizmadia, J. Mol. Struct. (Theochem) 467
(1999) 181.
[40] P.C. Chen, S.C. Chen, Int. J. Quantum Chem. 83 (2001) 332.
[41] M.W. Wong, Chem. Phys. Lett. 256 (1996) 391.
[42] G. Varsanyi, Vibrational Spectra of Benzene Derivatives,
Academic Press, New York, 1969.
[43] E.B. Wilson, Phys. Rev. 45 (1934) 706.
[44] M.A. Takipolsky, I.V. Tokmakov, V.A. Shlyapochnikov,
J. Mol. Struct. 510 (1999) 149.
[45] I.V. Tokmakov, V.A. Shlyapochnikov, Russ. Chem. Bull. 46
(1997) 1992.
J. Clarkson et al. / Journal of Molecular Structure 648 (2003) 203–214214](https://image.slidesharecdn.com/8da0d6e4-b5a6-47f9-9d24-1a27c0011592-150225131813-conversion-gate01/75/A-theoretical-study-of-the-structure-and-vibrations-of-2-4-6-trinitrotoluene-12-2048.jpg)

This document describes a theoretical study using density functional theory (DFT) to investigate the structure and vibrations of 2,4,6-trinitrotoluene (TNT). Two minimum energy structures of TNT were found, which differ by internal rotations of functional groups. Structure A has the lowest energy and geometry closest to crystal structures of TNT. DFT calculations at the B3LYP/6-311+G** level were able to accurately predict vibrational couplings and isotopic shifts observed in infrared and Raman spectra of solid TNT and isotopomers, while lower level calculations were not as accurate.



















































