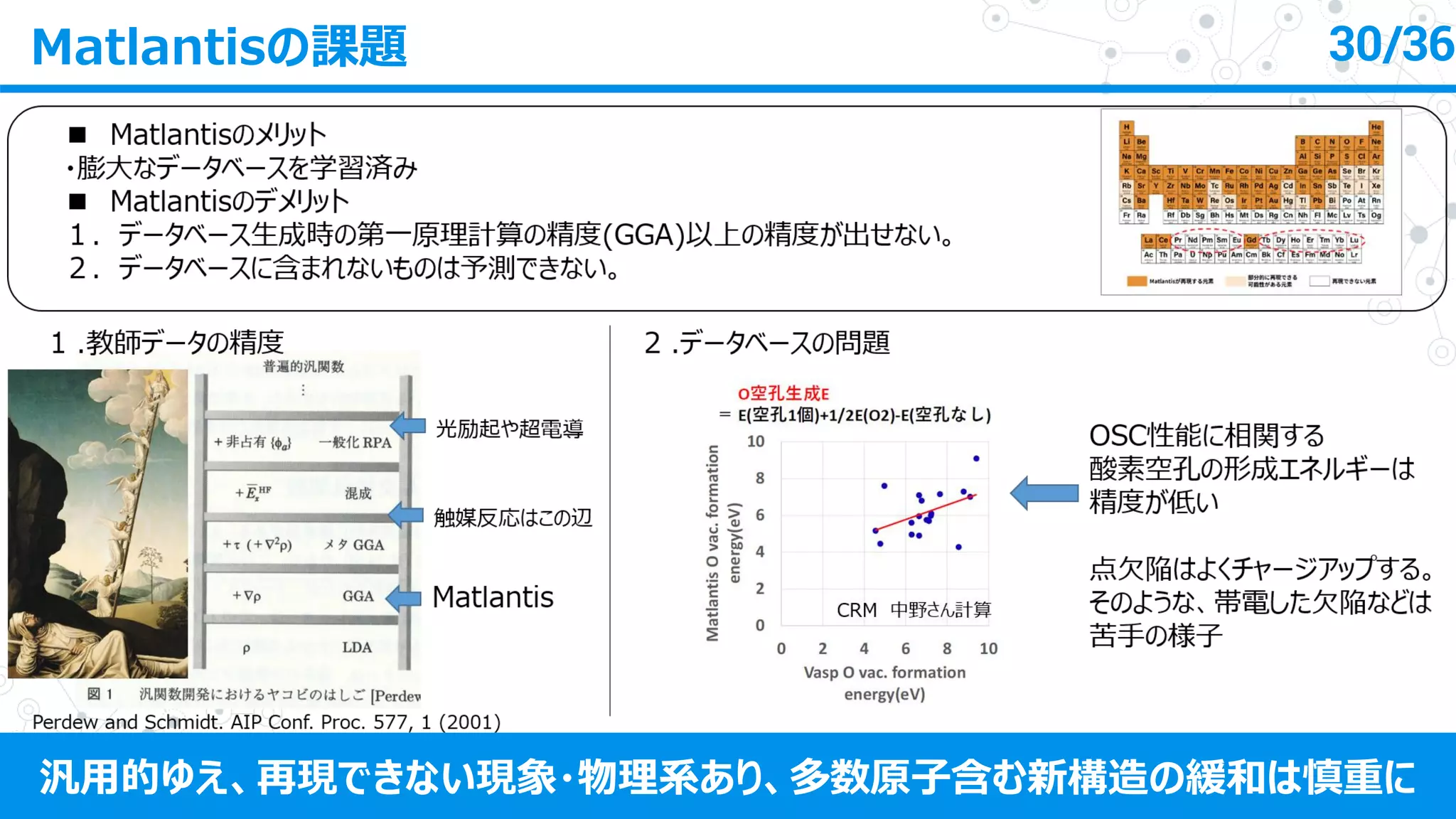
日本化学会第103春季年会(2023)におけるトヨタ自動車株式会社 山﨑久嗣様の発表資料です。 発表タイトル:機械学習によるハイスループット 第一原理計算の代替の可能性 https://matlantis.com/ja/






















![32
ベンチマーク 32/36
Formation
Energy(eV/atom)
Band Gap(eV) Bulk Moduli Shear Moduli Activation
Energy(eV)
CGCNN 0.031 0.292 0.047 0.099 0.392(LiCuOX our
941 structures)
CGCNN+3
(our unpublished results
with PFN) ‘19
0.038 0.361 0.164 0.284 0.143(COD data,
12,000 structures)
SchNet 0.033 0.345 0.066 0.099
MEGNET 0.030 0.307 0.060 0.099
GATGNN 0.033 0.280 0.045 0.075
ALIGNN 0.022 0.218 0.051 0.078
M3GNET 0.035(newer MP) 0.183(newer
MP)
0.058(newer
MP)
0.086(newer
MP)
Matlantis 0.0075(COD data
610,000)
- - - -
Matformer 0.021 0.211 0.043 0.073
Test MAE on Materials Project dataset
-2018.6.1 dataset, which contains 69239 crystals
*arXiv:2209.11807v1 [cs.LG] 23 Sep 2022](https://image.slidesharecdn.com/20230323-230612062531-0fcf1419/75/_-_20230323-32-2048.jpg)


















![[DL輪読会]近年のエネルギーベースモデルの進展](https://cdn.slidesharecdn.com/ss_thumbnails/energybasedmodel-200124020855-thumbnail.jpg?width=640&height=640&fit=bounds)














![[DL輪読会]GLIDE: Guided Language to Image Diffusion for Generation and Editing](https://cdn.slidesharecdn.com/ss_thumbnails/glide2-220107030326-thumbnail.jpg?width=640&height=640&fit=bounds)



![[DL輪読会]Inverse Design of Solid-State Materials via a Continuous Representation](https://cdn.slidesharecdn.com/ss_thumbnails/200214dl-200214034842-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DL輪読会]マテリアルズインフォマティクスにおける深層学習の応用](https://cdn.slidesharecdn.com/ss_thumbnails/181207dlwakasugipanasonicver4-181207003725-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DL輪読会]Learning to Simulate Complex Physics with Graph Networks](https://cdn.slidesharecdn.com/ss_thumbnails/learningtosimulatecomplexphysicswithgraphnetworks-200508054213-thumbnail.jpg?width=640&height=640&fit=bounds)

















