Download as ODP, PPTX


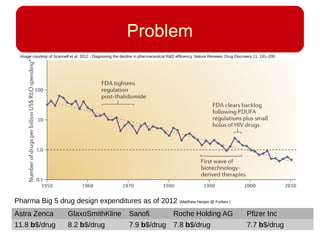
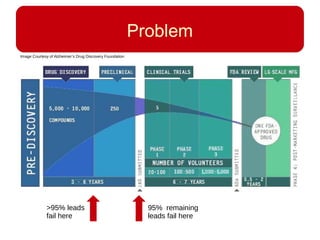
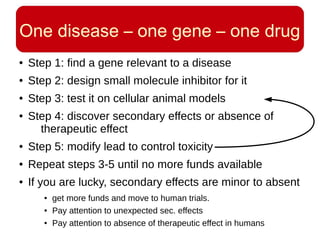


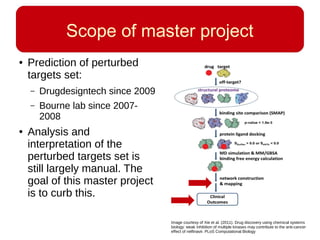


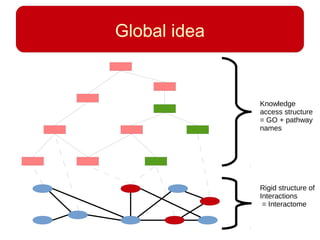
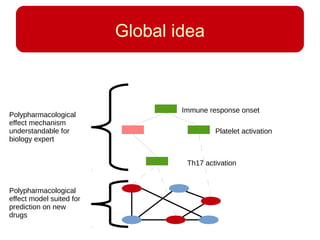

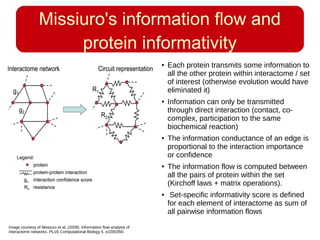
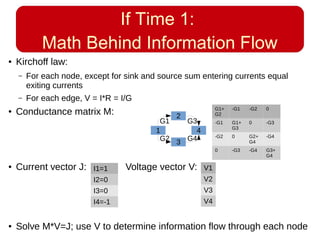
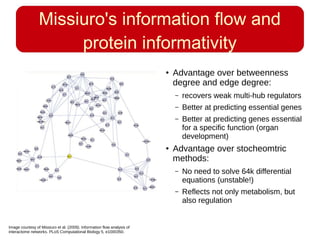
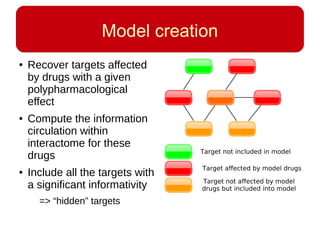
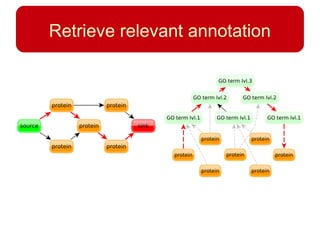

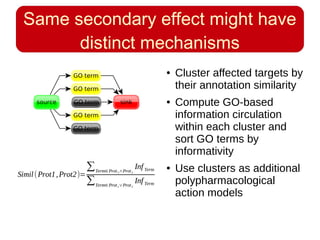
























This document discusses using systems biology approaches to predict and explain off-target drug effects. It notes that unexpected secondary drug effects and lack of therapeutic effects are major reasons drug development fails. The document proposes using computational methods to predict all protein targets a drug may affect and using systems biology to model the consequences, including secondary effects, unexpected therapeutic effects via drug repositioning, and unexpected lack of effects. It outlines a master's project to develop models of polypharmacological drug effects by analyzing networks of drug-affected protein targets and retrieving relevant biological annotation to interpret effects.



































































