Apidays New York 2024 - Accelerating FinTech Innovation by Vasa Krishnan, Fin...
Slideshow
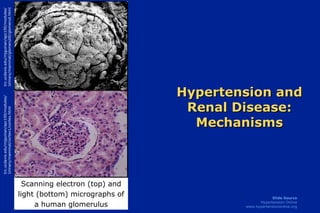
1. Hypertension and Renal Disease: Mechanisms trc.ucdavis.edu/mjguinan/apc100/modules/ Urinary/mammal/cortex1/cortex.html trc.ucdavis.edu/mjguinan/apc100/modules/ Urinary/mammal/glomeruli0/glomeruli.html Scanning electron (top) and light (bottom) micrographs of a human glomerulus
5. Effects of Vasodilators in the Normal Kidney L-Arginine NO eNOS (-) (-) L-Citrulline EDHF(s) Pgl 2 (-) (-) PMN M Platelet (-) VSMC EC
6. Imbalance in Factors Affecting Vascular Tone and Structure Nephron destruction and renal failure EDHF= endothelium-derived hyperpolarizing factors ROS= reactive oxygen species EDCF= endothelium-derived constricting factors Angiotensin II Catecholamines Endothelin-1 ROS Cytokines EDCF Nitric Oxide Prostacyclin Bradykinin EDHF Constrictors/ Growth Promoters Dilators/ Growth Inhibitors Vascular tone and structure
7. ROS Reduces the Biological Effects of NO + = OONO _ (-) Afferent Arteriole L-Arginine NO eNOS L-Citrulline NE VSMC PMN M Fibroblast EC Mast cell (+) O 2 •
8. Renin-Angiotensin Cascade Angiotensinogen Angiotensin I Angiotensin II AT 1 AT 2 AT n Bradykinin Inactive peptides Non-renin (eg tPA) Non-ACE (eg chymase) ACE Renin
9. Angiotensin II (Ang II) generated in the afferent arteriole interacts with AT 1 receptors on cellular components of the nephron Angiotensinogen Ang I Renin ACE Ang II AT 1 R = AT 1 Receptor
10.
11.
12.
13. Oxidative Stress: Endothelial Dysfunction and CAD/Renal Risk Factors O 2 Endothelial Cells and H 2 O 2 Vascular Smooth Muscle Endothelial Dysfunction Apoptosis Vasoconstriction Leukocyte adhesion Lipid deposition Thrombosis VSMC growth Hypertension Smoking Diabetes LDL Homocysteine Estrogen deficiency
14. Pivotal Role of ROS in Stimulus-Induced EC and VSMC Growth, Survival, and Apoptosis PDGF, Thrombin, Norepinephrine, Ang II, TNF, Ox-LDL, High Glucose, VEGF ROS Arachidonate Metabolism Mitochondrial Electron Transport Chain Cytochrome P 450 NOX-1 Oxidase Xanthine Oxidase Growth or Hypertrophy Survival Apoptosis Caspases NF- B Akt ERKs JNKs SAPKs p 38 MAPK Potential Targets of ROS Sources of ROS Growth/Death Survival Signals
15. Pathologic Processes Leading to Glomerular Injury and Proteinuria Ang II Increased glomerular pressure Ang II Urinary protein Glucose AGEs Glycoxidation (glycation) Efferent arteriolar constriction =angiotensin AT 1 receptor
16. Fibrosis and Nephron Loss: A Renal Response to Injury Vascular and/or Tubular Injury Glomerular cells Tubular cells Lymphocytes Macrophages Fibroblasts TGF- ET-1 CTGF Ang II PAI-1 PDGF bFGF TNF- IL-1 FIBROSIS
17. TGF- TGF- plays a key role in extracellular matrix formation in mesangium and interstitium that leads to fibrosis and loss of nephron units
18. bFGF PDGF Ang II TSP1 TGF- O 2 • TGF- plays a key role in extracellular matrix formation in mesangium and interstitium that leads to fibrosis and loss of nephron units O 2 •
19. TIMP bFGF PDGF Ang II Proteases (-) (-) (+) (+) (+) TSP1 ET-1 PAI-1 O 2 • TGF- TGF- plays a key role in extracellular matrix formation in mesangium and interstitium that leads to fibrosis and loss of nephron units O 2 •
20. Angiotensin II: Role in Renal Injury Angiotensin II AT 1 R AT 2 R NF- B TNFR1 TNFR2 Angiotensinogen Fibroblasts Proliferation and differentiation Matrix FIBROSIS Inflammation Cellular adhesion molecules Tubule cells TNF- + + Profibrotic cytokines
21. Aldosterone Promotes Renal Fibrosis by Multiple Mechanisms Adrenal Vascular Aldosterone PAI-1 Nitric oxide synthesis Na + influx into VSMC Norepinephrine uptake into VSMC Angiotensin II AT1R binding of Ang II Stimulates Inhibits Fibroblast collagen synthesis
22. Pathways Leading To Progressive Renal Failure Renal growth factor & cytokine activation Fibrogenesis Systemic hypertension Progressive Loss of Filtration Surface Area GFR Renal injury Nephron mass Glomerular hypertension Renal scarring Hyperlipidemia Filtration of plasma proteins (Proteinuria) Proximal tubule protein uptake Renal microvascular injury Influx of monocytes and macrophages Transdifferentiation of renal cells to fibroblast phenotype Brenner BM, Keane WF. 2001.
Editor's Notes
Hypertension and Renal Disease: Mechanisms The functional integrity of the kidney is vital to the maintenance of cardiovascular homeostasis. The nephron, comprised of the glomerulus (shown in the scanning electron photomicrograph on the top left and in the center of the light micrograph on the bottom left) and the tubules (surrounding the glomerulus in the light micrograph on the bottom left), is the primary functional unit of the kidney. The kidney plays a critical role in the long-term regulation of blood pressure. Thus, pathological abnormalities primary to the kidney may lead to an elevation of blood pressure. As a corollary, hypertension due to non-renal causes can damage the kidney. The resulting loss of renal mass, in turn, can secondarily lead to further elevations in blood pressure. This module summarizes some of the mechanisms that are thought to link hypertension with abnormal renal function. Photo Sources: Scanning electron micrograph (top): trc.ucdavis.edu/mjguinan/apc100/modules/Urinary/mammal/glomeruli0/glomeruli.html Light Photomicrograph (bottom): trc.ucdavis.edu/mjguinan/apc100/modules/Urinary/mammal/cortex1/cortex.html
Components of the Normal Nephron The architecture of the single nephron units that comprise the kidney facilitates interaction of its perfusion and filtration components and of the molecules produced by each of these components. A rich innervation of both the vascular and tubular structures of the nephron provides the conduit by which the kidney sends and receives signals from the brain. Cells in all layers of the arterioles (intima, media and adventitia), in the tubules and in the interstitium have the capacity to produce molecules that can modulate arteriolar tone, tubular reabsorptive function, amount and composition of the extracellular and mesangial matrix, and growth or replication of arteriolar and tubular cellular components. Differentiated vascular smooth muscle cells in the afferent arteriole, the juxtaglomerular cells, can produce renin in response to mechanical, chemical and neuronal stimuli. Some stimuli, such as stretch, originate in the systemic circulation as changes in arterial pressure while other stimuli, such as transcellular sodium flux, are produced by changes in tubular reabsorptive function. The close proximity of modified distal tubular cells, the macula densa, to the afferent arteriole and to the glomerulus facilitates transfer of these signals. The mesangial cells and their surrounding non-cellular matrix, the mesangial matrix, provide a structure to support the delicate glomerular capillaries. These cells also produce or transport molecules such as cytokines and growth factors that affect glomerular and tubular function.
Mechanisms of Renal Damage in HTN The kidney can be both a contributor to and a target of hypertension. Systemic blood pressure elevation is associated with increased vascular tone due, in part, to decreased production or actuin of vasodilator molecules, such as endothelium-derived nitric oxide and prostacyclin, at the same time as there is either maintenance or increased production of vasoconstrictors, such as angiotensin II and endothelin-1. This imbalance is often termed endothelial dysfunction. Increased renal sympathetic nerve activity may contribute to vasoconstriction and augment tubular reabsorption of sodium. Many of the molecules promoting vasoconstriction also cause long-term effects on the nephron by promoting growth of vascular smooth muscle cells and mesangial cells, decreasing apoptosis, stimulating chemotaxis of vascular inflammatory cells into the vessel wall and the tubular interstitium, and stimulating the production of molecules that expand and alter the composition of the extracellular matrix in the mesangium and interstitium. The resulting glomerular hypertension can lead to glomerular basement membrane damage and proteinuria. Other conditions, such as diabetes mellitus, may contribute to the glomerular basement membrane damage caused by hypertension or may induce it independently through alteration of basement membrane proteins. Both glomerular hypertension and proteinuria are associated with oxidative stress that can cause activation of circulating leukocytes and their diapedesis into the vessel wall and interstitium, can stimulate the release of cytokines and growth factors that leads to extracellular matrix formation, progressive sclerosis of both the glomerulus and tubules, and ultimately loss of nephron units. When the compensatory capacity of the remaining nephrons is exceeded, renal function progressively deteriorates and renal failure develops.
Consequences of Renal Damage in HTN This slide graphically illustrates the deleterious consequences of glomerular hypertension and proteinuria on a single nephron. Function changes that result from hypertension include a decline in glomerular filtration rate (GFR) and abnormalities in tubular function, including new onset or worsening of proteinuria. These functional changes lead to structural changes in the glomerular basement, expansion of the mesangial and interstitial matrix, ultimately resulting in sclerosis of both glomerular and tubular elements. More detail about the mechanisms by which this process is thought to occur can be found in the description of the previous slide.
Effects of Vasodilators in the Normal Kidney The vascular endothelium (EC) under normal conditions is an important source of vasodilators such as nitric oxide (NO), prostacyclin (PgI 2 ) and one or more endothelium-derived hyperpolarizing factors (EDHFs) that are not yet chemically characterized [cellular source of molecule identified by blue box and arrows]. Endothelium-derived NO has many functions [cellular targets identified by the orange arrows]. Among them are: vascular smooth muscle cell (VSMC) relaxation; modulation of renal medullary blood flow; inhibition of VSMC growth; inhibition of platelet aggregation; inhibition of polymorphonuclear (PMN) leukocyte and monocyte (M ) adhesion molecule expression and immigration into the vascular wall and interstitium; and augmentation of apoptosis or programmed cell death. The formation or the actions of endothelium-derived NO are blunted in patients with hypertension. This impairment may be more exaggerated in patients with concomitant diabetes mellitus. NO shares many of its actions in the vasculature and in the kidney with PgI 2, 20-HETE or other EDHFs. References: Vanhoutte PM and Boulanger CM. Endothelium-dependent responses in hypertension. Hypertens Res 1995;18:87-98. Lüscher TF and Bock HA. The endothelial L-arginine/nitric oxide pathway and the renal circulation. Klin Wochenschr 1991;69:603-609. Llinas MT, Gonzalez JD, Rodriguez F, Nava E, Taddei S, and Salazar FJ. Renal changes induced by nitric oxide and prostaglandin synthesis reduction: effects of trandolapril and verapamil. Hypertension 1998;31(2):657-664. Zou AP, Wu F, and Cowley AW, Jr. Protective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulation. Hypertension 1998;31(part 2):271-276. Stec DE, Mattson DL, and Roman RJ. Inhibition of renal outer medullary 20-HETE production produces hypertension in Lewis rats. Hypertension 1997;29 (part 2):315-319.
Imbalance in Factors Affecting Vascular Tone and Structure Hypertension is associated with an altered balance in the elaboration or biological action of vasodilator and vasoconstrictor molecules. The production of vasodilators like prostacyclin and nitric oxide is diminished in hypertension while the production of catecholamines, reactive oxygen species (ROS), angiotensin II, endothelin-1 and other endothelium-derived constricting factors is either maintained or increased. Many of the molecules that augment vascular tone also have longer-term mitogenic effects on vascular smooth muscle and glomerular mesangial cells, activate adhesion molecules on leukocytes and platelets with resulting influx of these cells into the vessel wall, decrease apoptosis, and stimulate extracellular (interstitial and mesangial) matrix formation, whereas molecules that promote vasodilation tend to inhibit these processes. References: Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int 2000;57[Suppl 75]:S2-S6. Asahi K, Ichimori K, Nakazawa H, Izuhara Y, Inagi R, Watanabe T, Miyata T, and Kurokawa K. Nitric oxide inhibits the formation of advanced glycation end products. Kidney Int 2000;58:1780-1787. Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000; 57[Suppl 75]:S7-14. Tharaux PL, Chatziantoniou C, Casellas D, Fouassier L, Ardaillou R, and Dussaule JC. Vascular endothelin-1 gene expression and synthesis and effect on renal type I collagen synthesis and nephroangiosclerosis during nitric oxide synthase inhibition in rats. Circulation 1999;99:2185-2191. Leehey DJ, Singh AK, Alavi N, and Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int 2000;58[Suppl 77]:S93-S98. Llinas MT, Gonzalez JD, Rodriguez F, Nava E, Taddei S, and Salazar FJ. Renal changes induced by nitric oxide and prostaglandin synthesis reduction: effects of trandolapril and verapamil. Hypertension 1998;31:657-664.
ROS Reduces the Biological Effects of NO The potent vasodilator molecule, nitric oxide (NO), is produced by constitutively expressed endothelial cell (EC) nitric oxide synthase (eNOS) from l-arginine via a 5 electron redox reaction [cell source illustrated by blue box and arrows]. NO also inhibits vascular smooth muscle cell (VSMC) proliferation and migration,mononuclear (M ) and polymorphonuclear (PMN) leukocyte adhesion molecule expression [illustrated by orange arrows], and platelet aggregation. Injured vascular smooth muscle cells or endothelial cells, and activated vascular wall mast cells, fibroblasts, macrophages, and leukocytes, as well as oxidation of norepinephrine (NE) from renal sympathetic nerves, produce increased amounts of reactive oxygen species (ROS) [illustrated by green box and arrows] that then interact with NO to form the potent cytotoxic peroxynitrite radical (OONO - ). This radical interacts with proteins in the kidney that are important for normal glomerular and tubular function to reduce their activity. References: Szilvassy Z, Csont T, Pali T, Droy-Lefaix MT, and Ferdinandy P. Nitric oxide, peroxynitrite and cGMP in atherosclerosis-induced hypertension in rabbits: beneficial effects of cicletanine. J Vasc Res 2001;38:39-46. Thuraisingham RC, Nott CA, Dodd SM, and Yaqoob MM. Increased nitrotyrosine staining in kidneys from patients with diabetic nephropathy. Kidney Int 2000;57:1968-1972. Walker LM, Walker PD, Imam SZ, Ali SF, and Mayeux PR. Evidence for peroxynitrite formation in renal ischemia-reperfusion injury: studies with the inducible nitric oxide synthase inhibitor L- N(6)-(1-Iminoethyl)lysine. J Pharmacol Exp Ther 2000;295:417-422.
Renin-Angiotensin Cascade This slide shows the renin-angiotensin cascade. There are at least two alternative pathways for angiotensin II formation that do no rely on either renin or angiotensin converting enzyme (ACE). In the non-renin pathway, tissue plasminogen activator (tPA) forms angiotensinsin II directly from angiotensinogen, bypassing the renin-mediated production of angiotensin I as an intermediate. A second alternative pathway involves enzymes like chymase that can form angiotensin II from angiotensin I via an ACE-independent mechanism. These alternative pathways are implicated in the gradual return toward pre-treatment angiotensin II concentrations during treatment of patients with ACE inhibitors, and provide a rationale for considering angiotensin receptor blockers (ARBs) that directly inhibit the binding of angiotensin II to the AT 1 receptor either in conjunction with or as an alternative to ACE inhibitor therapy. References: Balcells E, Meng QC, Johnson WH, Jr., Oparil S, and Dell'Italia LJ. Angiotensin II formation from ACE and chymase in human and animal hearts: methods and species considerations. Am J Physiol 1997;273(4 Pt 2):H1769-H1774. Petrie MC, Padmanabhan N, McDonald JE, Hillier C, Connell JM, and McMurray JJ. Angiotensin converting enzyme (ACE) and non-ACE dependent angiotensin II generation in resistance arteries from patients with heart failure and coronary heart disease. J Am Coll Cardiol 2001;37:1056-1061.
Angiotensin II Angiotensin I (Ang I) formed in the afferent arteriole via the action of renin synthesized and secreted by the modified vascular smooth muscle cells (VSMC) in the arteriolar tunica media (juxtaglomerular cells) then interacts with angiotensin converting enzyme, a membrane bound enzyme on the luminal plasma membrane of the endothelial cell to form angiotensin II (Ang II). Ang II can bind to and activate angiotensin AT 1 receptors located on VSMCs, endothelial cells, tubular cells and mesangial cells. Binding of angiotensin II to afferent arteriolar juxtaglomerular cell AT 1 receptor diminishes further renin production, thus providing an intrarenal feedback loop for the control of renin secretion and angiotensin II generation.
Role of Angiotensin II in Chronic Renal Disease Angiotensin II plays a pivotal role in pathological processes in hypertension that ultimately leads to renal glomerular and tubular destruction and renal failure. Angiotensin II, acting either through signal transducing mechanisms (top left box) or directly on cells to stimulate the production or activation of mediators (top right box) causes infiltration of inflammatory cells, increased production of mesangial and interstitial matrix with resultant glomerular and tubular injury and destruction (nephron loss). The remaining normal nephrons are forced to compensate by increasing filtration rate which, in the presence of increased angiotensin II, increases glomerular capillary pressure and perpetuates this progressive spiral of deteriorating renal function. Abbreviations: TGF- , transforming growth factor-beta, CTGF, connective tissue growth factor, PAI-1, plasminogen activator inhibitor-1
Angiotensin II Induces Oxidative Stress in the Kidney Angiotensin II stimulates several key enzymes or cytokines in vascular and tubular cells within the kidney to produce increased amounts of reactive oxygen species (ROS). Selected key enzymes whose activation leads to increased ROS formation are listed on this slide. In addition, angiotensin II activates NF- B, a nuclear factor pivotal to the production of inflammatory cytokines. These cytokines exert their effect, in part, through oxidative mechanisms. References: Griendling KK, Sorescu D, and Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86:494-501. Haugen EN, Croatt AJ, and Nath KA. Angiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int 2000;58:144-152. Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57 [Suppl75]:S7-14.
Renal Sources of ROS There are several key enzymes or cytokines in vascular and tubular cells within the kidney that produce increased amounts of reactive oxygen species (ROS). Selected key enzymes whose activation leads to increased ROS formation are listed on this slide. Superoxide anion is spontaneously converted to hydrogen peroxide (H 2 O 2 ), a reaction that is facilitated in the presence of superoxide dismutase. Hydrogen peroxide is then converted to water and divalent oxyen by catalase. References: Griendling KK, Sorescu D, and Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86:494-501. Haugen EN, Croatt AJ, and Nath KA. Angiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int 2000;58:144-152. Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57[Suppl75]:S7-14.
Oxidative Stress: Endothelial Dysfunction and CAD/Renal Risk Factors A variety of conditions or insults listed at the top of the illustration that are known risk factors for either coronary artery disease (CAD), progressive renal insufficiency, or both, adversely affect endothelial cell or vascular smooth muscle cell function by increasing the formation of reactive oxygen species such as superoxide anion and hydrogen peroxide. The resultant reduction in the actions of endothelium-derived vasodilators/growth inhibitors such as prostacyclin and nitric oxide with maintenance or increased formation of endothelium-derived vasoconstrictors/growth promoters, such as angiotensin II, endothelin-1, and PAI-1, has significant vascular and renal pathophysiological consequences. Some of the mechanisms by which progressive coronary and renal injury occur include increased apoptosis or programmed cell death that contributes to vascular wall remodeling, activation of cell adhesion molecules resulting in adherence of both mononuclear and polymorphonuclear leukocytes to the vascular wall with subsequent infiltration, deposition of oxidized lipids in the vessel wall, vasoconstriction, both hypertrophy and hyperplasia of vascular smooth muscle cells, and a propensity for thrombus formation.
Pivotal Role of ROS in Stimulus-Induced EC and VSMC Growth, Survival, and Apoptosis Reactive oxygen species (ROS) serve an important role as signal transduction molecules that stimulate or inhibit a variety of vascular and endothelial cell processes that lead to vascular remodeling. Key stimuli, listed at the top of the slide produce ROS via multiple pathways, several of which are illustrated here. These ROS, in turn, activate intracellular kinase, NF- B, or caspase signaling pathways in vascular smooth muscle and endothelial cells that regulate hypertrophy or proliferation, cell survival or programmed cell death (apoptosis). References: Irani K. Oxidant signaling in vascular cell growth, death, and survival : a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res 2000;87(3):179-83. Griendling KK, Sorescu D, and Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86(5):494-501.
Pathologic Processes Leading to Glomular Injury and Proteinuria When Ang II is increased, greater AT 1 receptor-mediated constriction of efferent than afferent arterioles increases single nephron glomerular filtration rate and raises intraglomerular pressure, causing glomerular hypertension. Sustained or severe increases in intraglomerular pressure can lead to glomerular basement membrane damage, glomerular endothelial dysfunction, and ultimately, extravasation of protein into Bowman’s capsule. In addition to hypertension, conditions like diabetes that are associated with increased oxidative stress (increased formation of reactive oxygen species) independent of hypertension and glyco-oxidative modification of proteins (advanced glycation endproducts or AGEs) comprising the glomerular basement membrane can lead to extravasation of protein.
Fibrosis and Nephron Loss: A Renal Response to Injury Angiotensin II stimulates both mesangial and renal tubular cells to produce transforming growth factor- (TGF- ). TGF- stimulates increased production of extracellular matrix proteins. Angiotensin II also stimulates the production of endothelin-1 (ET-1) from the endothelial cells of the arterioles. ET-1, in turn, also stimulates increased production of mesangial and interstitial matrix proteins. This process leads to loss of cellular elements, injury to renal tubules, fibrosis, and ultimately, loss of the entire nephron. In diabetics, increased glucose, via an increase in angiotensinogen, facilitates the production of more angiotensin II. References: Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol 2000;15(3-4):290-301.
TGF- Angiotensin II and Thrombospondin (TSP1) can both stimulate the production of transforming growth factor-b (TGF- ) by tubuloepithelial cells and fibroblasts. TGF- , in turn, causes proliferation of fibroblasts and tubuloepithelial cells. TGF- ultimately increases extracellular matrix proteins, likely by several mechanisms. 1) TGF- stimulates production of several growth factors including basis fibroblast growth factor (bFGF) and platelet derived growth factor (PDGF) that stimulate the formation of extracellular matrix (ECM) proteins. 2) TGF- also has a direct effect on ECM protein expression. 3) TGF- stimulates the formation of the receptors to which ECM proteins adhere. 4) TGF- increases the expression of tissue inhibitors of metalloproteases (TIMP) that, in turn, prevents proteases from degrading ECM proteins. 5) TGF- stimulates the endothelial cell production of endothelin-1 (ET-1). The effects of ET-1 are illustrated on a separate slide. References: Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57[Suppl75]:S7-14. Inoki K, Haneda M, Ishida T, Mori H, Maeda S, Koya D, Sugimoto T, and Kikkawa R. Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-beta in mesangial cells. Kidney Int 2000;58[Suppl 77]:76-80.
TGF- Angiotensin II and Thrombospondin (TSP1) can both stimulate the production of transforming growth factor-b (TGF- ) by tubuloepithelial cells and fibroblasts. TGF- , in turn, causes proliferation of fibroblasts and tubuloepithelial cells. TGF- ultimately increases extracellular matrix proteins, likely by several mechanisms. 1) TGF- stimulates production of several growth factors including basis fibroblast growth factor (bFGF) and platelet derived growth factor (PDGF) that stimulate the formation of extracellular matrix (ECM) proteins. 2) TGF- also has a direct effect on ECM protein expression. 3) TGF- stimulates the formation of the receptors to which ECM proteins adhere. 4) TGF- increases the expression of tissue inhibitors of metalloproteases (TIMP) that, in turn, prevents proteases from degrading ECM proteins. 5) TGF- stimulates the endothelial cell production of endothelin-1 (ET-1). The effects of ET-1 are illustrated on a separate slide. References: Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57[Suppl75]:S7-14. Inoki K, Haneda M, Ishida T, Mori H, Maeda S, Koya D, Sugimoto T, and Kikkawa R. Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-beta in mesangial cells. Kidney Int 2000;58[Suppl 77]:76-80.
TGF- Angiotensin II and Thrombospondin (TSP1) can both stimulate the production of transforming growth factor-b (TGF- ) by tubuloepithelial cells and fibroblasts. TGF- , in turn, causes proliferation of fibroblasts and tubuloepithelial cells. TGF- ultimately increases extracellular matrix proteins, likely by several mechanisms. 1) TGF- stimulates production of several growth factors including basis fibroblast growth factor (bFGF) and platelet derived growth factor (PDGF) that stimulate the formation of extracellular matrix (ECM) proteins. 2) TGF- also has a direct effect on ECM protein expression. 3) TGF- stimulates the formation of the receptors to which ECM proteins adhere. 4) TGF- increases the expression of tissue inhibitors of metalloproteases (TIMP) that, in turn, prevents proteases from degrading ECM proteins. 5) TGF- stimulates the endothelial cell production of endothelin-1 (ET-1). The effects of ET-1 are illustrated on a separate slide. References: Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57[Suppl75]:S7-14. Inoki K, Haneda M, Ishida T, Mori H, Maeda S, Koya D, Sugimoto T, and Kikkawa R. Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-beta in mesangial cells. Kidney Int 2000;58[Suppl 77]:76-80.
Angiotensin II: Role in Renal Injury This simplified schema depicts a likely mechanism by which angiotensin II promotes renal glomerulosclerosis and tubulo-interstitial fibrosis. Additional details and explanation can be found on the preceding slide. References: Klahr S and Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000;57[Suppl 75]:S7-14.
Aldosterone Promotes Renal Fibrosis by Multiple Mechanisms The potent mineralocorticoid, aldosterone, is produced by the adrenal zona glomerulosa in response to angiotensin II stimulation. There also is evidence that the myocardium as well as vascular endothelial and smooth muscle cells are capable of synthesizing aldosterone. Aldosterone has been implicated in vascular, myocardial and renal fibrosis that occurs in patients with arteriosclerosis, congestive heart failure and renal failure, particularly if hypertension is also present and is accompanied by activation of the RAAS. The mechanisms by which aldosterone contributes to vascular and renal fibrosis is not well understood, but there are several experimental findings that support the mechanisms included in the illustration. Aldosterone has been reported to: 1) augment affinity of ang II for the AT 1 receptor, thereby enhancing the pressor effects of angiotensin II; 2) augment the influx of sodium into vascular smooth muscle cells, contributing to VSMC hypertrophy; 3) potentiate the action of norepinephrine by inhibiting its uptake into vascular smooth muscle cells; 4) contribute to endothelial dysfunction by inhibiting nitric oxide synthesis; 5) stimulate the production of PAI-1, which promotes fibrosis by mechanisms previously illustrated; and 6) promotes synthesis of Type I collagen by interstitial fibroblasts. References: Hatakeyama H, Miyamori L, Fujita T, Takeda Y, Takeda R, and Yamamoto H. Vascular aldosterone: Biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem 1994;289:24316-24320. Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, Moalic JM, and Swynghedauw B. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J Biol Chem 1998;273:4883-4891. Ullian ME, Schelling JR, and Linas S. Aldosterone enhances angiotensin II receptor binding and inositol phosphate responses. Hypertension 1992;20:67-73. Kornel L and Smoszna-Konaszewska B. Aldosterone (ALDO) increases transmembrane influx of Na+ in vascular smooth muscle (VSM) cells through increased synthesis of Na+ channels. Steroids 1995;60:114-119. Weber MA and Purdy RE. Catecholamine mediated constrictor effects of aldosterone on vascular smooth muscle. Life Sci 1982;30:2009-2017. Ikeda U, Kanbe T, Nakayama I, Kawahara Y, Yokoyama M, and Shimada K. Aldosterone inhibits nitric oxide synthesis in vascular smooth muscle cells induced by interleukin-1b. Eur J Pharmacol 1995;290:69-73. Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, Vaughan DE, and Fogo AB. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 2000;58:1219-1227. Epstein M. Aldosterone as a mediator of progressive renal disease: pathogenetic and clinical implications. Am J Kidney Dis 2001;37(4):677-88.
Pathways Leading to Progressive Renal Failure This slide summarizes the multiple mechanisms by which non-hypertensive and hypertensive insults lead to renal scarring that results in loss of nephrons and ultimately to renal failure. Both systemic hypertension and non-hypertensive injury that causes loss of single nephron units results in hypertension in the remaining glomeruli (glomerular hypertension). Glomerular hypertension can lead to injury to the glomerular basement membrane causing it to leak plasma proteins into the urine. Attempts by the proximal tubules to reabsorb this filtered protein causes injury to the tubular cells, activates an inflammatory response, and is associated with the development of lipid metabolic abnormalities that create further oxidative stress on the already compromised glomerulus. The resultant tubular inflammatory response and renal microvascular injury activate pathways that lead to fibrosis and scarring of both glomerular and tubular elements of the nephron. An additional consequence of glomerular hypertension and resultant reduction in glomerular filtration rate (GFR) activates growth factors and cytokines that promote an influx of monocytes and macrophages into the vessel wall and into the renal interstitium, and also causes the differentiation of renal cells into fibroblasts. Monocytes, macrophages and fibroblasts are capable of producing those growth factors and cytokines that activate pathways leading to expansion of extracellular matrix, fibrosis and loss of both tubular and glomerular structures.